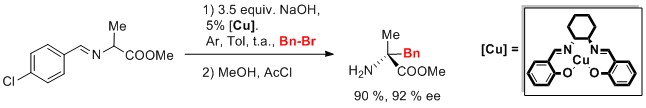
[47] Rh(I) complexes with hemilabile thioether-functionalized NHC ligands as catalysts for [2+2+2] cycloaddition of 1,5-bisallenes and alkynes accepted in ACS. Catalysis 2023
Jordi, V.; Miquel, S.; Achard, T.; Bellemin-Laponnaz, S.; Pla-Quintana, A.; Roglans, A.
[46] A stable and photoreactive copper-iodide cubane suitable for direct post-functionalization.
Bissessar, D.; Egly, J.; Achard, T.; Steffanut, P.; Mauro, M.; Bellemin-Laponnaz, S.
Eur. J. Inorg. Chem. 2022, e202200101.
Tetranuclear copper-iodide cluster consisting of a {Cu4I4} cubane and diphenylphosphine ligand has been found as a highly stable precursor for further post-functionalization through hydrophosphination reaction. UV irradiation of the cluster in the presence of various alkenes allows direct functionalization of the latter, giving access to a wide range of copper cubane complexes, a source of molecular diversity for luminescent materials. Such a strategy provided access to the first example of water-soluble luminescent cubane copper iodide.
Highlighted in Chemistry Views 5th May 2022
A patent application has been filed on these results : D. Bissessar, S. Bellemin-Laponnaz, P. Steffanut, Tetra-nuclear neutral copper (I) complexes with diaryl phosphine ligands, US2021253611A1.

[45] Half-sandwich ruthenium complexes bearing hemilabile κ2-(C,S)-thioether-functionalized NHC ligands: application to amide synthesis from alcohol and amine.
Egly J., Chen W., Maisse-François A., Bellemin-Laponnaz S., Achard T.
Eur. J. Inorg. Chem. 2022, e202101033 (early access)
Amide synthesis is one of the most crucial transformations in chemistry and biology. Among various catalytic systems, N-heterocyclic carbene (NHC)-based ruthenium (Ru) catalyst systems have been proven to be active for direct synthesis of amides by sustainable acceptorless dehydrogenative Coupling of primary alcohols with amines. Most often, these catalytic systems usually use monodentate NHC and thus require an additional ligand to obtain high reactivity and selectivity. In this work, a series of cationic Ru(II)(η6–p-cymene) complexes with thioether-functionalized N-heterocyclic carbene ligands (imidazole and benzimidazole-based) have been prepared and fully characterized. These complexes have then been used in the amidation reaction and the most promising one (i. e. 3 c) has been applied on a large range of substrates. High conversions albeit with moderate yields have generally been obtained.

[44] Synthesis, Structural Characterization and Antiproliferative Activity of Gold(I) and Gold(III) Complexes Bearing Thioether-Functionalized N-Heterocyclic Carbenes.
De Marco, R.; Dal Grande, M.; Baron, M.; Orian, L.; Graiff, C.; Achard, T.; Bellemin-Laponnaz, S.; Pöthig, A.; Tubaro, C.
Eur. J. Inorg. Chem. 2021, 40, 4196.
Gold(I,III) complexes with N-heterocyclic carbene ligands having a thioether side-arm were synthesized and fully characterized both in solution and in the solid state; they displayed moderate in vitro antiproliferative properties, without a significant influence by the gold oxidation state.

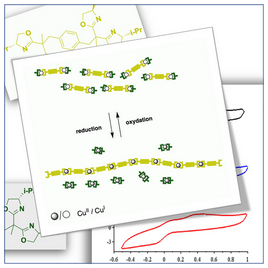
[43] Polymerization/depolymerization of chiral metallo-supramolecular assembly induced by redox change.
Marinova, M.; Bonnefont, A.; Achard, T.; maisse-François, A.; Bellemin-Laponnaz, S.
We report on the polymerization/depolymerization of chiral metallo-supramolecular assembly by CuI/CuII redox change. By combining a monotopic enantiopure ligand with a ditopic ligand of opposite configuration, ML2-type complexes are generated with chiral self-recognition or self-discrimination depending on the oxidation state of copper. In presence of CuI, the formation of heterochiral complexes is favored, thus generating dinuclear species whereas CuII advocates for the formation of homochiral species, namely, a mixture of mononuclear species and metallo-supramolecular polymeric species. Thus, cyclic voltammetry was used to study such a chirality-induced stimulus sensitive polymerization/depolymerization process.
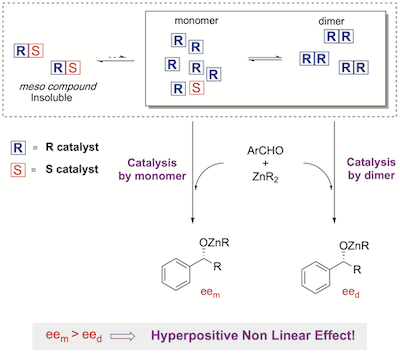
[42] Absence of Non-Linear Effects despite evidence for catalyst aggregation.
Geiger, Y.; Achard, T.; Maisse-François, A.; Bellemin-Laponnaz, S.
One might think that the absence of a non-linear effect (NLE) might necessarily mean that only monomeric complexes catalyze a studied enantioselective reaction. Herein, a counter-example using an ephedrine-derived ligand in asymmetric alkylzinc additions is shown. The system is catalyzed by both monomers and dimers, although no NLE is apparent.
ChemRxiv 2021 Preprint, doi.org/10.26434/chemrxiv.13911659.v1

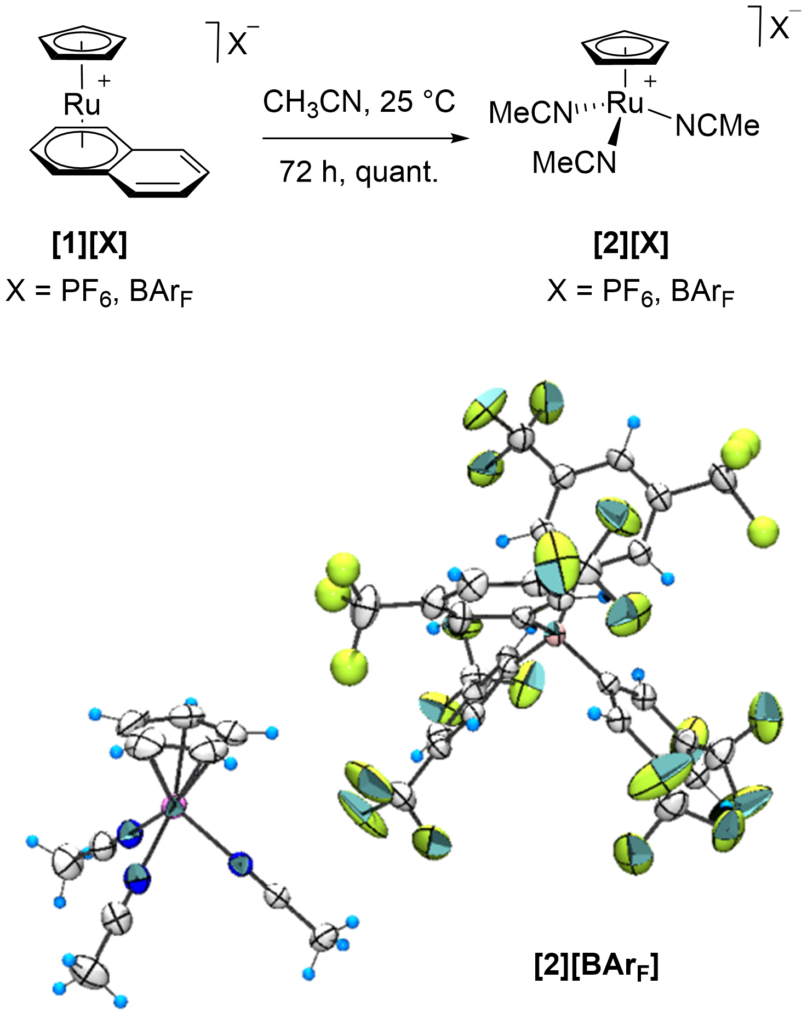
[41] Preparation and structural characterization of [CpRu(1,10-phenanthroline)(CH3CN)][X] and precursor complexes (X= PF6, BArF, TRISPHAT-N)
Achard,T.; Léo Egger, L.; Tortoreto, C.; Guénée L.; Lacour, J.
Helv. Chim. Acta 2020, 103, e2000190
Cationic [Ru(η5-C5H5)(CH3CN)3]+ complex, tris(acetonitrile)(cyclopentadienyl)ruthenium(II), gives rise to a very rich organometallic chemistry. Combined with diimine ligands, and 1,10-phenanthroline in particular, this system efficiently catalyzes diazo decomposition processes to generate metal-carbenes which undergo a series of original transformations in the presence of Lewis basic substrates. Herein, syntheses and characterizations of [CpRu(Phen)(L)] complexes with (large) lipophilic non-coordinating (PF6− and BArF−) and coordinating N-TRISPHAT− anions are reported.
[40] Recent advances on catalytic osmium-free olefin syn-dihydroxylation
Achard , T.; Bellemin-Laponnaz, S
syn‐1,2‐Diols are valuable scaffolds for building a vast number of biologically active compounds and are also important synthetic intermediates in organic synthesis. This review focuses on a concise summary of the main metal‐catalyzed methods of olefin syn‐dihydroxylation developed over the past ten years to circumvent the use of osmium.

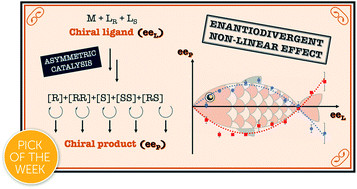
[39] Hyperpositive non-linear effects: enantiodivergence and modelling.
Geiger, Y.; Achard, T.; Maisse-François, A.; Bellemin-Laponnaz, S.
Highlighted in Nature Chemistry Reviews
Unmasking unusual behaviour 5th November 2020
The chiral ligand N-methylephedrine (NME) was found to catalyse the addition of dimethylzinc to benzaldehyde in an enantiodivergent way, with a monomeric and a homochiral dimeric complex both catalysing the reaction at a steady state and giving opposite product enantiomers. A change in the sign of the enantiomeric product was thus possible by simply varying the catalyst loading or the ligand ee, giving rise to an enantiodivergent non-linear effect. Simulations using a mathematical model confirmed the possibility of such behaviour and showed that this can lead to situations where a reaction gives racemic products, although the system is composed only of highly enantioselective individual catalysts. Furthermore, depending on the dimer’s degree of participation in the catalytic conversion, enantiodivergence may or may not be observed experimentally, which raises questions about the possibility of enantiodivergence in other monomer/dimer-catalysed systems.
[38] Copper(I) complexes with remotely functionalized phosphine ligands: synthesis, structural variety, photophysics and effect onto the thermally-activated delayed fluorescence.
Egly, J.; Bissessar, D.; Achard, T.; Steffanut, P.; Mauro, M.; Bellemin-Laponnaz, S.
Inorg. Chim. Acta (Special Issue) 2021, 514, 119971
The synthesis, chemical and photophysical investigation of a series of eight novel copper-halide derivatives with different nuclearity is herein presented. One mononuclear copper(I) complex with formula [CuI(pyridine)(P)2], where P is a functionalized phosphine, six dinuclear derivatives bearing the rhombic {Cu2I2} subunits and with general formula [Cu(μ-I)(P)(N)]2 (N = pyridine, quinoline and 4-cyanopyridine) as well as one tetranuclear copper-iodide clusters of general formula [Cu(μ-I)P]4 consisting of a cubane-like {Cu4X4} motif were straightforwardly prepared, also by means of mechano-chemical synthetic procedure. The phosphine ligands were prepared in excellent yield by using simple hydrophosphination reactions. For five of the investigated cuprous iodide derivatives their atom connectivity and solid-state crystalline packing was unambiguously confirmed by solving their single-crystal X-ray structure. All the investigated complexes resulted into photo- and thermally-stable luminescent species. In the solid state as microcrystalline powder samples, the complexes display intense photoluminescence ranging from the sky-blue to orange-red portion of the spectrum with PLQY values of 0.28–0.50 and 0.34–0.97 at room and low (77 K) temperature, respectively, depending on the nature of both the coordinated N-heteroaromatic ring and phosphine ligands. In spite of the remote location of the functionalization of the phosphine, profound effects are observed onto the solid-state emission properties highlighting the importance of microenvironment for the emitters in the aggregated phase

[37] N-Heterocyclic Carbene Platinum(IV) as metallodrug candidates: synthesis and 195Pt NMR chemical shift trend.
Bouché, M.; Vincent, B.; Achard, T.; Bellemin-Laponnaz, S.
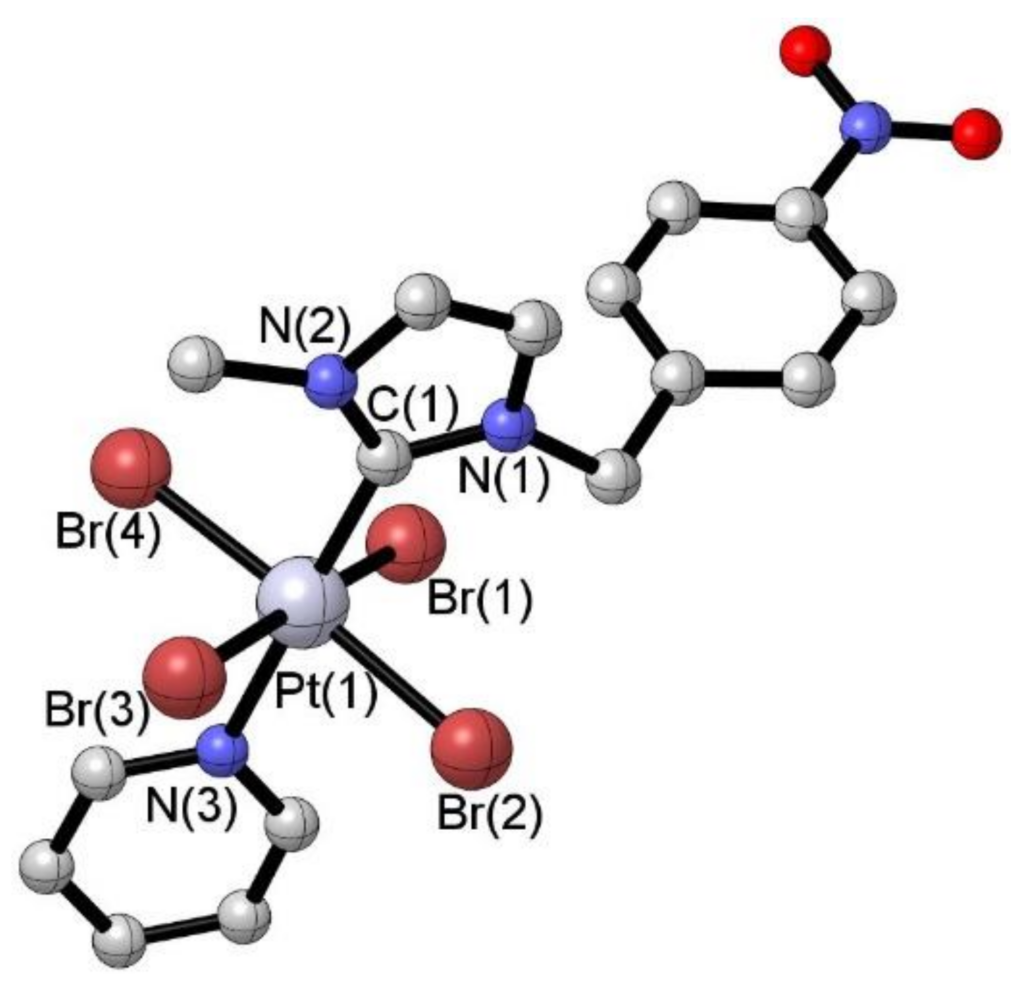
[36] Chiral stimuli-responsive metallo-supramolecular assembly induced by Cu(II)/Cu(I) change.
Marinova, M.; Bonnefont, A.; Achard, T.; Maisse-François, A.; Bellemin-Laponnaz, S.
Chem. Commun. 2020, 56, 8703-8706.
We investigated the selective formation of homoleptic and heteroleptic metal complexes controlled by means of the chiral molecular instruction of the ligand and the coordination geometry of the metal. Our results showed that chiral self-recognition or self-discrimination may be induced by the CuI/CuII redox transition using cyclic voltammetry. The further use of chiral ditopic ligands led to metallo-supramolecular copolymers with stimuli-responsive controlled arrangement
[35] Observation of hyperpositive non-linear effect in catalytic asymmetric organozinc additions to aldehydes.
Geiger, Y.; Achard, T.; Maisse-François, A.; Bellemin-Laponnaz, S.
Chirality (Special Issue) 2020, doi.org/10.1002/chir.23271.
Asymmetric amplification is a phenomenon that is believed to play a key role in the emergence of homochirality in life. In asymmetric catalysis, theoretical and experimental models have been investigated to provide an understanding of how chiral amplification is possible, in particular based on non‐linear effects. Interestingly, it has been proposed a quarter century ago that chiral catalysts, when not enantiopure might even be more enantioselective than their enantiopure counterparts. We show here that such hyperpositive non‐linear effect in asymmetric catalysis is indeed possible. An in‐depth study into the underlying mechanism was carried out, and the scheme we derive differs from the previous proposed models.
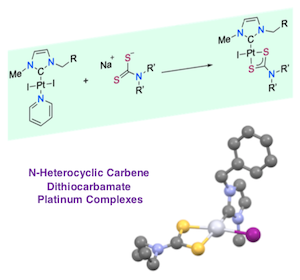
[34] Synthesis and characterization of N-heterocyclic carbene dithiocarbamate platinum complexes with antitumoral activity.
Verron, R.; Achard, T.; Seguin, C.; Fournel, S.; Bellemin-Laponnaz, S.
Eur. J. Inorg. Chem. 2020, 26, 2552
A series of platinum(II) complexes bearing N‐heterocyclic carbene NHC of the type [(NHC)PtX(L)] where L is a dithiocarbamate ligand and X iodide were prepared and fully characterized. The molecular structure of one derivative was determined by single‐crystal X‐ray analyses confirming that the dithiocarbamate is acting as a bidentate ligand to the metal. All complexes demonstrated potent in vitro antiproliferative activities against cancer cells. Mechanistic investigations revealed that mitochondrial dysfunction and ROS (Reactive Oxygen Species) production were correlated with the cytotoxic process induced by these complexes.
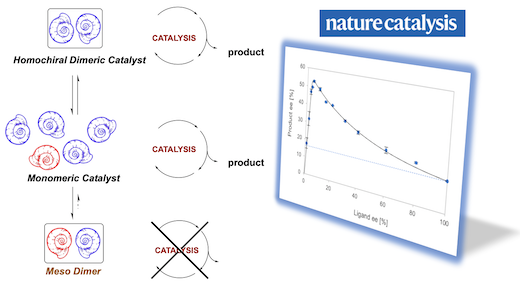
[33] Observation of hyperpositive non-linear effects in asymmetric catalysis.
Geiger, Y.; Achard, T.; Maisse-François, A.; Bellemin-Laponnaz, S.
Nature Catalysis 2020, 3, 422-426.
Highlighted in Chemistry World 25th March 2020
Highlighted in University of Strasbourg News 20th April 2020
Highlighted in CNRS INC News 9th June 2020
Asymmetric amplification is a curious phenomenon that is believed to play a key role in the emergence of biological homochirality, and thus of life itself. In asymmetric catalysis, it is achieved via positive nonlinear effects, which allow high product enantiomeric excesses with a non-enantiopure catalyst. However, it has also been proposed that non-enantiopure catalysts may be even more enantioselective than their enantiopure counterparts, although such a case has never been experimentally observed so far. Here, we present an example of such a hyperpositive nonlinear effect in asymmetric catalysis. We found that addition of dialkylzinc reagents to benzaldehyde gave higher product enantiomeric excesses with only partially resolved chiral N-benzyl-ephedrine ligands. A mechanistic study was carried out and our results point towards a two-component catalysis, where mononuclear as well as aggregated catalysts are in equilibrium and in competition. These results introduce an unprecedented class of asymmetric amplification in enantioselective catalysis.
[32] Synthesis, characterization, catalytic and biological application of half-sandwich ruthenium complexes bearing hemilabile (κ2–C,S)-thioether-functionalised NHC ligands.
Chen, W.; Egly, J.; Poblador-Bahamonde, A. I. I.; Maisse-François, A.; Bellemin-Laponnaz, S.; Achard, T.
Dalton Trans. 2020, 49, 3243 – 3252
A series of cationic Ru(II)(η6-p-cymene) complexes with thioether-functionalised N-heterocyclic carbene ligands have been prepared and fully characterized. Steric and electronic influence of the R thioether substituent on the coordination of the sulfur atom was investigated. The molecular structure of three of them has been determined by means of X-ray diffractrometry and confirmed the bidentate (κ2-C,S) coordination mode of the ligand. Interestingly, only a single diastereomer, as an enantiomeric couple, was observed in the solid state for complexes 1c, 1i and 1j. DFT calculations established a low energy inversion barrier between the two diastereomers through a sulfur pyramidal inversion pathway with R donating group while a dissociative/associative mechanism is more likely with R substituents that contain electron withdrawing group, thus suggesting that the only species observed by the 1H-NMR correspond to an average resonance position of a fluxional mixtures of isomers. All these complexes were found to catalyse the oxydant-free double dehydrogenation of primary amine into nitrile. Finally, preliminary results of the biological effects on various human cancer cells of four selected Ru complexes are reported
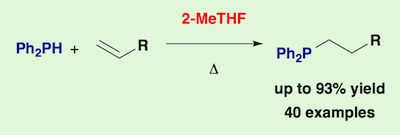
[31] Catalyst-free hydrophosphination of alkenes in presence of 2-methyltetrahydrofuran: a green and easy access to a wide range of tertiary phosphines.
Bissessar, D.; Egly, J.; Achard, T.; Steffanut, P.; Bellemin-Laponnaz, S.
A hydrophosphination reaction that is free of base, acid and catalyst, using only 2-methyltetrahydrofuran as additive has been performed. A new family of mono-, di-, tri- and tetra-phosphines compounds are obtained in good to excellent yields by adding diphenylphosphine to alkenes, mono- and polyfunctional acrylics (based on acrylate and methacrylate motifs) and acrylamide substrates. Addition of four equivalent of bio-mass derived 2-MeTHF into the reaction media improves both conversion and time of the reaction and reduces the sensitivity of the reactants over oxidation. This simple, straightforward and atom-economic method respects the principles of Green Chemistry. Furthermore, in each case this transformation shows an exclusive regioselectivity towards the anti-Markovnikov products.
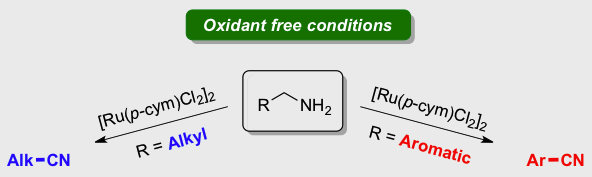
[30] Easy ruthenium‐catalysed oxidation of primary amines to nitriles under oxidant free conditions
Achard T., Egly, J., Sigrist, M., Maisse-François A., Bellemin-Laponnaz S.
Chem. Eur. J. 2019; 25, 13271-13274.
A dehydrogenation of primary amine to give the corresponding nitrile under oxidant‐ and base‐free conditions catalysed by simple [Ru(p‐cym)Cl2]2 with no extra ligand is reported. The system is highly selective for alkyl amine whereas benzylamine derivatives gave the nitrile product together with the imine in ratio ranging from 14:1 to 4:1 depending on the substrate. Preliminary mechanistic investigations have been performed to identify the key factors that govern the selectivity.
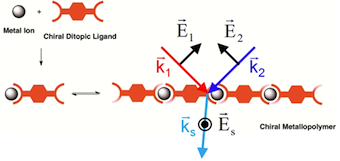
[29] Chiral Self-Sorting Process with Ditopic Ligands: Alternate or Block Metallopolymer Assembly as a Function of the Metal Ion
Maya Marinova M., Torres-Werlé M., Taupier G., Maisse-François A., Achard T., Boeglin B., Dorkenoo H.D., Bellemin-Laponnaz S.
ACS Omega 2019, 4, 2, 2676-2683
We report an extensive study on the coordination behavior of chiral ditopic bridging ligands, which lead to metallosupramolecular polymers in the presence of Zn(II) and Cu(II) in solution. With the help of UV–vis and circular dichroism spectroscopies, we show that the metallopolymer sequence can be controlled by chirality and by the choice of the metal ion. Although the formation of a block metallopolymer proceeds through the assembly of homoleptic complexes, an alternate metallopolymer may be obtained only when heteroleptic complexes are formed. This demonstrates how the prevalent coordination geometries at metal centers may be used to control the sequences of the metallopolymers.

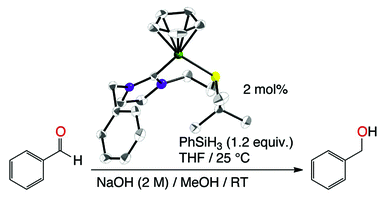
[28] Synthesis, characterization, and catalytic application in aldehyde hydrosilylation of half-sandwich nickel complexes bearing (κ1-C)- and hemilabile (κ2-C,S)-thioether-functionalised NHC ligands
Ulm F., Poblador-Bahamonde A., Choppin S., Bellemin-Laponnaz S., Chetcuti M., Achard T. and Ritleng V.
Dalton Trans. 2018, 47, 17134-17145
Neutral nickel–N-heterocyclic carbene complexes, (κ1–C)-[NiCpBr{R-NHC-(CH2)2SR′}] and cationic [NiCp{R-NHC-(CH2)2SR′}](PF6) complexes, which bear a N-bound thioether side arm, were prepared. A careful study by both NMR (1H, Variable Temperature) and solvent DFT calculation shows that for neutral complexes the thioether part is not coordinated to the metal center and that the observed disatereotopicity is more likely due to significant steric congestion. In contrast, cationic complexes shows displayed a C,S-chelation in the solid state as observed with X-ray crystallography analysis; the 1H NMR spectra (CDCl3, CD2Cl2, or thf-d8) at room temperature showed no diastereotopic NCH2CH2S protons, thus suggesting the possible displacement of the sulphur atom by the respective solvents and/or very fast sulphur inversion. DFT calculations established a low energy inversion process in all cases (+9 ≤ ΔG‡ ≤ +13 kcal mol−1) as well as a favourable solvent coordination process (ΔG‡ ≈ +11 kcal mol−1; ΔG ≈ −7 kcal mol−1) with a solvent such as THF, thus suggesting that sulphur inversion and/or solvent coordination can both account for the absence of diastereotopy at room temperature, depending on the solvent. While all complexes catalysed the hydrosilylation of benzaldehyde in the absence of any additive, the cationic C,S-chelated complexes 2 proved more active than the sterically constrained neutral species 1. In particular, 2c proved to be the most active pre-catalyst and its catalytic charge could be lowered down to 2 mol% with PhSiH3 as the hydrogen source.
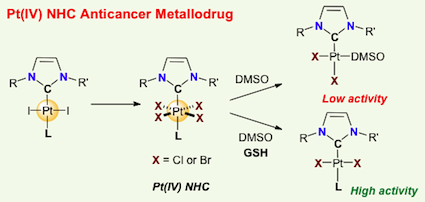
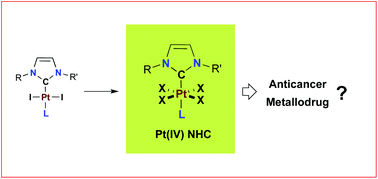
[27] Exploring Diversity in Platinum(IV) N-Heterocyclic Carbene Complexes: Synthesis, Characterization, Reactivity and Biological Evaluation.
Bouché, M.; Bonnefont, A.; Achard, T.; Bellemin-Laponnaz, S.
Platinum(IV) complexes stabilized by N-heterocyclic carbene ligands of the type [(NHC)PtX4L], where L is a neutral nitrogen-based ligand and X is a halide anion (Br, Cl), were prepared by using straightforward and high-yielding synthetic routes and the scope was extended to amphiphilic derivatives. The complexes demonstrated in vitro antiproliferative activities against several cancer cell lines. In particular, a representative Pt(IV) complex, namely, [(NHC)PtCl4(pyridine)], displayed efficient antiproliferative activity against cisplatin-resistant cancer cells.
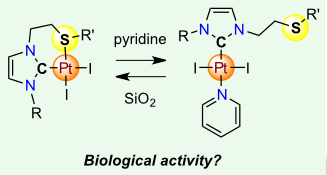
[26] Synthesis, structural characterisation and anti-proliferative activity of (κ1–C)- and (κ2–C,S)-Pt(II) complexes bearing thioether-functionalized N-heterocyclic carbenes
Egly, J.; Bouché, M.; Chen, W.; Maisse-François, A.; Achard, T.; Bellemin-Laponnaz, S.
Eur. J. Inorg. Chem. 2018, 159
Platinum(II) complexes bearing thioether‐functionalized N‐heterocyclic carbene ligands have been synthesized and characterized. The effect of the thioether on the biological activity of the platinum centre has been investigated on various cancer cell lines
[25] Optically Active Sum-Frequency Generation as an Advanced Tool for Chiral Metallopolymer
Taupier, G.; Torres-Werlé, M.; Boeglin, A.; Maisse-François, A.; Achard, T.; Bellemin-Laponnaz, S.; Dorkenoo, H. D.
[24] Synthesis and application of dynamic self-supported enantioselective catalysts.
Bellemin-Laponnaz, S.; Achard, T.; Bissessar, D.; Geiger, Y.; Maisse-François, A.
Coord. Chem. Rev. 2017, 332, 38.
Since chiral coordination complexes have a broad scope for asymmetric synthesis, their immobilization has been the subject of intensive research. Indeed, the immobilization of chiral catalysts is an appealing approach to solve problems such as product contamination or catalyst reuse. Numerous approaches have been developed and despite promising results each immobilization technique displays several strengths and limitations that make them highly specific. During the last years, dynamic self-supported catalysis has emerged as an alternative and new strategy to circumvent some limitations of classical immobilized catalysts. In such system, a metal-organic self-assembly is in equilibrium with discrete monomeric active species, a case that offers exciting opportunities for catalysis. Recent work in this area shows that these systems can compete with their homogeneous counterparts. This review highlights the recent developments in the growing field of dynamic self-supported asymmetric catalysis.

[23] Platinum(IV) N-heterocyclic carbene complexes: their synthesis, characterisation and cytotoxic activity
Bouché, M.; Dahm, G.; Wantz. M.; Fournel, S.; Achard, T.; Bellemin-Laponnaz, S.
Dalton Trans., 2016, 45, 11362.
Platinum(II) N-heterocyclic carbene complexes have been oxidized by bromine or iodobenzene dichloride to provide the fully characterised corresponding platinum(IV) NHC complexes. Antiproliferative activities of Pt(IV) NHC complexes were assayed against several cancer cell lines and the results were correlated with respect to their stability. Mechanistic investigations revealed that mitochondrial dysfunction and ROS production were associated with the cytotoxic process induced by these compounds
[22] Robust and Recyclable Self-Supported Chiral Nickel Catalyst for the Enantioselective Michael Addition.
Bissessar, D.; Achard, T.; Bellemin-Laponnaz, S.
Adv. Synth. Catal. 2016, 358, 1982
A simple chemical modification of a chiral diamine ligand may produce a robust and recyclable enantioselective catalyst. Metallopolymers based on chiral cyclohexyldiamine-containing ditopic ligands and nickel(II) complexes have been readily prepared and applied in catalytic enantioselective Michael additions of 1,3-dicarbonyl compounds to nitroalkenes. High yields and good enantioselectivities have been obtained and the catalytic systems have been recycled up to 11 times without loss of either activity or enantioselectivity at a low catalyst loading of 0.75 mol%. Moreover, the nickel metallopolymers were found to be air- and moisture-stable, which enabled this chemistry to be carried out on the bench without the use of any air-free techniques and with non-degassed solvents. Finally, the nature of the catalyst was studied by non-linear effect experiments, giving a negative non-linear effect (NLE) as a consequence of an in situ decrease in the ee of the active species consistent with the trapping of homochiral aggregates.

[21] Selective formation of cis N-Heterocyclic Carbene-Pt(II)-Pnictogen Complexes and In Vitro Evaluation of their Cytotoxic Activities Toward Cancer Cells.
Bouché, M.; Dahm, G.; Maisse-François, A.; Achard, T.; Bellemin-Laponnaz, S.
Eur. J. Inorg. Chem. 2016, 2828
A series of heterotopic platinum complexes combining N-heterocyclic carbene (NHC) and pnictogen-based ligands [Pn = P(nBu)3, PPh3, AsPh3 and SbPh3] has been synthesized and fully characterized. Use of an excess of Pn ligand led to the formation of cationic bis-Pn platinum-NHC complexes in high yield, with both Pn ligands being arranged trans to each other. Several representative complexes could be characterized by X-ray crystallographic studies thus confirming the ligand arrangement around the metal. This simple approach allows the generation of diversity in metallodrug candidates. Biological activities on various human cancer cells have been studied and compared with those of cisplatin. The results confirmed the high cytotoxicity of these NHC-platinum complexes bearing pnictogen ligands.

[20] Advances in Homogeneous Catalysis Using Secondary Phosphine Oxides (SPOs): Pre-ligands for Metal Complexes
Achard, T.
Special edition : The French Connection
The secondary phosphine oxides are known to exist in equilibrium between the pentavalent phosphine oxides (SPO) and the trivalent phosphinous acids (PA). This equilibrium can be displaced in favour of the trivalent tautomeric form upon coordination to late transition metals. This tutorial review provides the state of the art of the use of secondary phosphine oxides as pre-ligands in transition metal-catalysed reactions. Using a combination of SPOs and several metals such as Pd, Pt, Ru, Rh and Au, a series of effective and original transformations have been obtained and will be discussed here.

[19] Synthesis of Spiro Ketals, Orthoesters, and Orthocarbonates by CpRu-Catalyzed Decomposition of α-Diazo-β-ketoesters
Tortoreto, C.; Achard, T.; Egger, L.; Guénée, L.; Lacour, L.
Reactions of α-diazo-β-ketoesters with cyclic ketones, lactones, and carbonates are reported. Thanks to the combined use of salt [CpRu(CH3CN)3][BArF] and 1,10-phenanthroline as catalyst for the diazo decomposition, effective and practical syntheses of spiro bicyclic ketals, orthoesters, and orthocarbonates are afforded.

[18] Palladium-catalyzed [2+1] cycloadditions affording vinylidenecyclopropanes as precursors of 7-membered carbocycles
Lepronier, A.; Achard,T.; Giordano, L.; Tenaglia, A.; Buono, G.; Clavier, H.
Adv. Synth. Catal. 2016, 358, 631.
Palladium(II) acetate in association with secondary phosphine oxides provides an efficient catalytic system for [2+1] cycloadditions starting from oxanorbornene derivatives and tertiary propargyl esters giving rise to vinylidenecyclopropanes. This reaction is specific to bidentate phosphinito–phosphinous acid ligands generated from secondary phosphine oxides. Moreover, vinylidenecyclopropanes were straightforwardly converted into oxabicyclo[3.2.1]oct-2-ene derivatives through a palladium-catalyzed ring-expansion. Finally, the oxa bridge cleavage of oxatricyclic compounds yields functionalized 7-membered

[17] Regio- and Enantioselective Allylation of Phenols via Decarboxylative Allylic Etherification of Allyl Aryl Carbonates Catalyzed by (Cyclopentadienyl)ruthenium(II) Complexes and Pyridine-Hydrazone Ligands
Egger, L.; Tortoreto, C.; Achard, T.; Monge, D.; Ros, A.; Fernández, R.; Lassaletta, J. M.; Lacour, J.
Adv. Synth. Catal. 2015, 357, 3325.
(Cyclopentadienyl)tris(acetonitrile)ruthenium hexafluorophosphate [CpRu(CH3CN)3][PF6] in combination with pyridine-hydrazone ligands efficiently catalyzes the asymmetric decarboxylative allylic rearrangement of allyl aryl carbonates. Formation of C-O bonds with high regio- and enantioselectivity ratios (up to 95:5 and 98% ee) is obtained. Good stereocontrol of the pseudotetrahedral geometry of the CpRu moiety is achieved by the hydrazone ligand and its “electron-poor” nature is evidenced through the epimerization of the hexacoordinated TRISPHAT-N anion.

[16] Original Reactivity of α-Diazo-β-ketoesters Catalyzed by CpRu Complexes.
Tortoreto C., Achard T., Austeri M., Zeghida W., Lacour J.
Using α-diazo-β-ketoesters as reagents and combinations of CpRu fragments and diimine ligands as catalysts, a series of original transformations have been obtained that can be rationalized by the formation of metal carbenes and metal-bound ylide intermediates. Interesting 1,3-dioxole, enol-acetal and 1,4-dioxene motifs are obtained directly when the reactive mixture is reacted in presence of aldehydes or ketones, THF and epoxides.

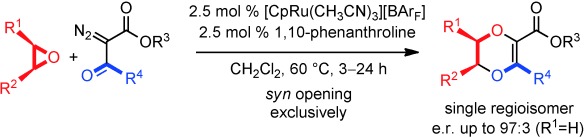
[15] CpRu-catalyzed carbene insertions into epoxides: 1,4-dioxene synthesis via SN1-like chemistry with retention of configuration.
Achard T., Tortoreto C., Poblador-Bahamonde A.I., Bürgi T., Guénée L., Lacour J.
Angew. Chem. Int. Ed. 2014, 53, 6140.
Rather than lead to the usual deoxygenation pathway, metal carbenes derived from α-diazo-β-ketoesters undergo three-atom insertions into epoxides using a combination of 1,10-phenanthroline and [CpRu(CH3CN)3][BArF] as the catalyst. Original 1,4-dioxene motifs are obtained as single regio- and stereoisomers. A perfect syn stereochemistry (retention, e.r. up to 97:3) is observed for the ring opening, which behaves as an SN1-like transformation.
This publication has been highlighted in SYNLETT 2014, 25(16), A136–A138.
[14] S-Streogenic Ligands in Asymmetric Hydrogenation. Neutral vs. Cationic Rhodium Complexes of Bulky N-Phosphino Sulfinamide Ligands.
Doran, S.; Achard, T.; Riera, A. and Verdaguer, X.
J. Organomet. Chem., 2012, 712, 135.
N-di-tert-butylphosphino-tert-butylsulfinamide coordinates to rhodium to form a neutral complex. The cationic rhodium species can be formed by protonation of the said complex and its structure was elucidated by X-ray analysis. The efficacies of the neutral and the cationic species were tested in the asymmetric hydrogenation of Z-MAC.

[13] Enol-Acetal Synthesis via Carbenoid C-H Insertions into Tetrahydrofurans Catalyzed by CpRu Complexes.
Tortoreto C.; Achard,T.; Zeghida W.; Austeri M.; Guenée L.; Lacour J.
Angew. Chem. Int. Ed. 2012, 51, 5847.
Intermolecular C-O instead of C-C bond formation is achieved with [CpRu(CH3CN)3][PF6] and diimine ligands as catalysts of the decomposition of α-diazo-β-ketoesters in THF leading to original products of 1,3 C-H insertion

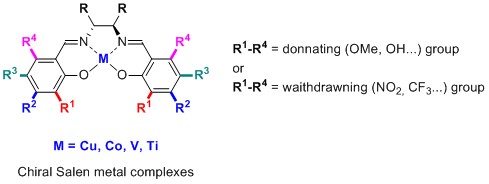
[12] Chiral salen ligands designed to form polymetallic complexes
Achard,T.R.J.; Clegg, W.; Harrington, R.W.; North, M.
Chiral salen ligands capable of forming polymetallic complexes have been designed. The ligands possess substituents in the 4,4′-positions, but have no substituent in the 3,3′-positions to allow a second metal ion access to the salen oxygen atoms. All of the complexes were tested as asymmetric phase transfer catalysts for the asymmetric alkylation of an alanine methyl ester, forming (R)-α-methyl phenylalanine methyl ester with up to 85% ee.

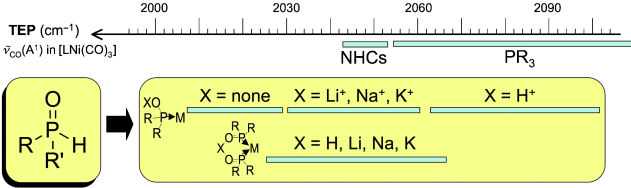
[11] Assessment of the highly modular electronic properties of P-ligands stemming from secondary phosphine oxides.
Martin, D.; Moraleda, D.; Achard, T.; Giordano L.; Buono, G.
Chem. Eur. J. 2011, 17, 12729.
We report the study of the net donating ability of monodentate and bidentate P ligands stemming from secondary phosphine oxides (SPOs). We experimentally measured and/or calculated the frequencies of CO stretching modes of various metal carbonyl complexes. The inferred electronic properties of the ligands span an unprecedented range, going from π-accepting phosphite-like compounds, to extremely electron-donating ligands outclassing N-heterocyclic carbenes.
[10] N-Benzyl-N-phosphino-tert-butylsulfinamide and Its Coordination Modes with Ir (I), Cu(I), Pd(II) and Pt(II): P,S or P,O?
Achard, T.; Benet-Buchholz, J.; Escudero-Adan, E.; Riera, A.; Verdaguer, X.
Organometallics 2011, 30, 3119.
Here we studied the coordination mode of optically pure N-benzyl-N-phosphino-tert-butylsulfinamide (1a) toward Ir(I), Cu(I), Pd(II), and Pt(II). Ligand 1a can work as a hemilabile bidentate P,O or P,S or monodentate P ligand depending on the nature of the metal and its electronic and steric environment.

[9] A Regio- and Diastereoselectivite Platinum-Catalyzed Tandem [2+1]/[3+2] Cycloaddition Sequence.
Achard, T.; Lepronier, A.; Gimbert, Y. ; Clavier, H. ; Giordano L.; Tenaglia, A. ; Buono, G.
Angew. Chem. Int. Ed. 2011, 50, 3552.
Platinum complexes bearing phosphinous acids have efficiently promoted a tandem intermolecular sequence of [2+1]/[3+2] cycloaddition reactions. This process gave access to novel tricyclic systems and the cascade reactions were regio- and diastereoselective (see scheme; Cy=cyclohexyl). The [3+2] cycloaddition reaction was investigated further and two different alkyne partners were used

[8] N-Phosphino sulfinamide compounds as efficient ligands in rhodium(I) catalyzed [2+2+2] cycloaddition reactions
Brun, S. ; Parera, M. ; Pla-Quintana, A. ; Roglans, A.; León, T. ; Achard, T. ; Solà, J. ; Verdaguer, X. ; Riera, A.
The combination of cationic rhodium(I) complexes with N-phosphino tert-butylsulfinamides (PNSO) ligands is efficient for catalytic intra- and intermolecular [2+2+2] cycloaddition reactions. PNSO ligands are a new class of chiral bidentate ligands, which have the characteristic of combining the easily accessible sulfur chirality with the coordinating capacity of phosphorous. Cycloaddition of open-chained and macrocyclic E-enediynes with these chiral complexes have proved to be highly efficient in terms of yields, giving moderate enantiomeric excesses of the corresponding cyclohexadiene derivatives. In addition Rh(I)/PNSO complexes catalyzed the intermolecular cycloaddition of diynes with monoalkynes in mild reaction conditions and short reaction times

[7] Steric Control in the Synthesis of Phosphinous Acid-Coordinated Mono- and Binuclear Platinum(II) Complexes
Achard, T.; Giordano L.; Tenaglia, A.; Gimbert, Y. ; Buono, G.
Organometallics 2010, 29, 3936.
Several phosphinous acid-coordinated platinum complexes were prepared and characterized. In the presence of secondary phosphine oxide (2 equiv), PtCl2(cod) was converted to a series of cis/trans platinum complexes PtCl2[R1R2POH]2 featuring phosphinous acids (PAs) as ligands (20−29). A balance between the steric and electronic effects of ligands governs their coordination mode.

[6] Cationic Rhodium (I) Complexes of N-Phosphino-tert-butylsulfinamide Ligands: Synthesis, Structure, and Coordination Modes.
Achard, T.; Benet-Buchholz, J.; Riera, A.; Verdaguer, X.
Organometallics, 2009, 28, 480.
N-Phosphino-tert-butylsulfinamide ligands (PNSO) can work as either P,O or P,S chelating ligands when attached to the square planar rhodium center. Complexes bearing diene ligands such as [Rh(PNSO)(NBD)][TfO] and [Rh(PNSO)(COD)][TfO] provided P,O coordination, whereas [Rh(PNSO)2][TfO]-type complexes provided P,S coordination.

[5] N-Phosphino-p-tolylsulfinamide Ligands: Synthesis, Stability, and Application to the Intermolecular Pauson−Khand Reaction
Reves, M.; Achard, T.; Sola, J.; Riera, A.; Verdaguer, X.
J. Org. Chem., 2008, 73, 7080.
Here we synthesized a family of racemic and optically pure N-phosphino-p-tolylsulfinamide (PNSO) ligands. Their stability and coordination behavior toward dicobalt−alkyne complexes was evaluated. The resulting optically pure major complexes were tested in the asymmetric intermolecular Pauson−Khand reaction and yielded up to 94% ee.

[4] Enantio- and Diastereoselective Darzens condensations
Achard, T.; Belokon, Y.; Ilyin, M.; Moskalenko, M.; Pizzato, F.; North, M.
Tetrahedron Letters 2006, 48, 2965.
A cobalt(salen) complex has been shown to catalyse the asymmetric Darzens condensation between α-haloamides and aldehydes, allowing both the relative and absolute stereochemistry of the epoxy-amide products to be controlled. Under optimal conditions, cis-epoxides can be obtained diastereoselectively with up to 50% enantiomeric excess, whilst by changing the leaving group and base, trans-epoxides can be produced diastereoselectively with up to 43% enantiomeric excess.

[3] Diastereoselective Darzens condensations.
Achard, T.; Belokon, Y.; Hunt, J.; Pizzato, F.; North, M.
Tetrahedron Letters 2006, 48, 2961.
N,N-Diphenyl-α-haloacetamides undergo Darzens condensations with aldehydes under heterogeneous reaction conditions in the presence of a metal hydroxide base. By appropriate choice of solvent, base and halide, very high diastereoselectivities favouring formation of the cis– or trans-epoxide can be obtained.

[2] Asymmetric Catalysis of Carbon-Carbon Bond-Forming Reactions Using Metal(salen) Complexes”
Achard, T.; Clutterbuck, L.M.; North, M.
The use of metal(salen) complexes in the asymmetric syntheses of cyanohydrins and amino acids have been developed as important and reliable methods of obtaining these highly versatile chiral intermediates.
[1] Influence of aromatic substituents on metal(II)salen catalysed, asymmetric synthesis of α-methyl α-amino acids
Achard, T.; Belokon, Y.; Fuentes, J.A.; Parsons, T.; North, M.
The influence of substituents on both the aromatic rings of the catalyst, and the benzylidene unit of the substrate are investigated in the (salen)copper(II) catalysed asymmetric benzylation of alanine derivatives.