D1. Mécanismes de relaxation dans les systèmes moléculaires
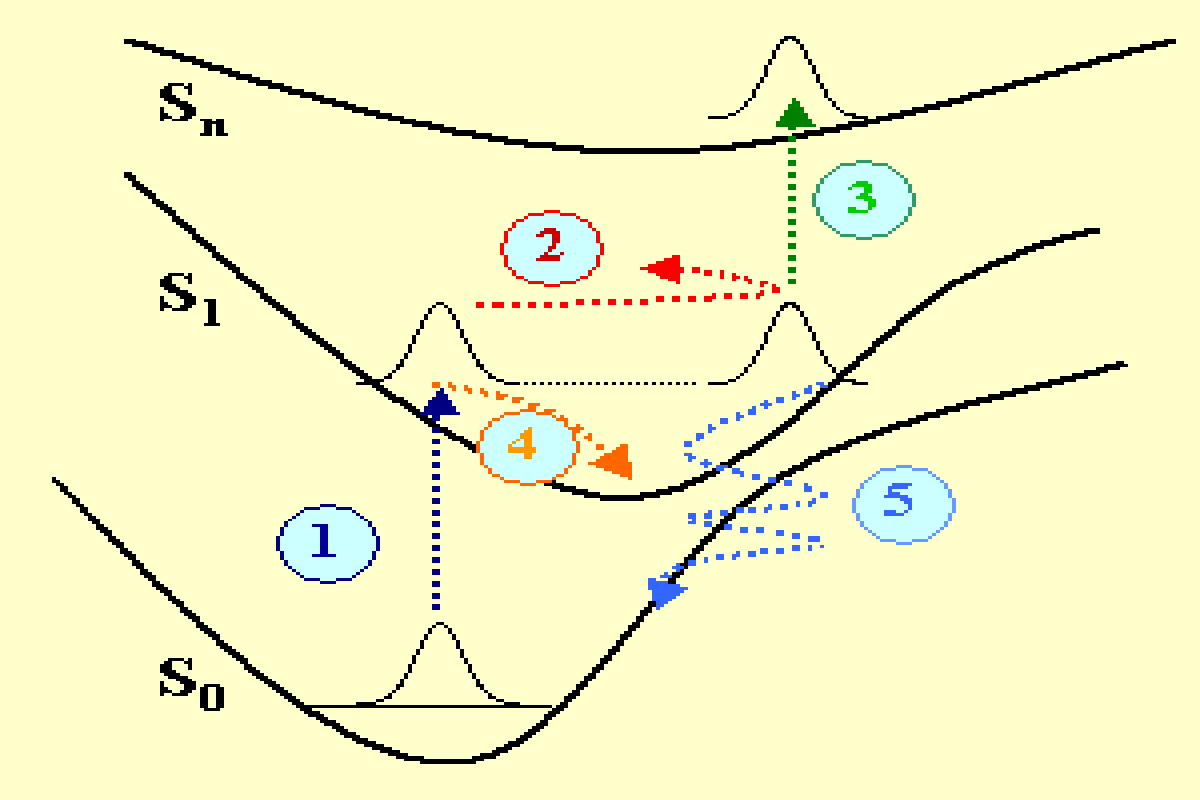
Les réactions chimiques ou les transformations photo-physiques des molécules ont leur origine dans les processus physiques élémentaires qui se produisent aux échelles du nanomètre et de la femtoseconde. Ces processus dépendent fortement de la dynamique moléculaire qui peut être observée en utilisant les techniques de la spectroscopie optique femtoseconde. Sur la figure 1 plusieurs mécanismes physiques sont décrits dans un potentiel représenté dans un espace de configuration simplifié. Une molécule possédant trois états électroniques singulets S0, S1, Sn, est excitée avec une impulsion laser ultra-brève de quelques femtosecondes de durée. La transition optique initiale depuis l’état fondamental vers l’état excité S1 (mécanisme 1 en bleu foncé) contribue à exciter une superposition cohérente d’états, c’est à dire un paquet d’ondes qui est hors équilibre et se déplace en oscillant sur la surface de potentiel (mécanisme 2 en rouge). Un paquet d’ondes est également excité dans l’état fondamental (non représenté). L’énergie et la période du paquet d’ondes vibrationnel dépendent bien entendu des vibrations moléculaires particulières qui sont excitées (élongation, torsion, rotation des liaisons chimiques) ainsi que de la forme spécifique des surfaces de potentiel dans l’espace de configuration (potentiels connectés Frank-Condon, anharmonicité…).
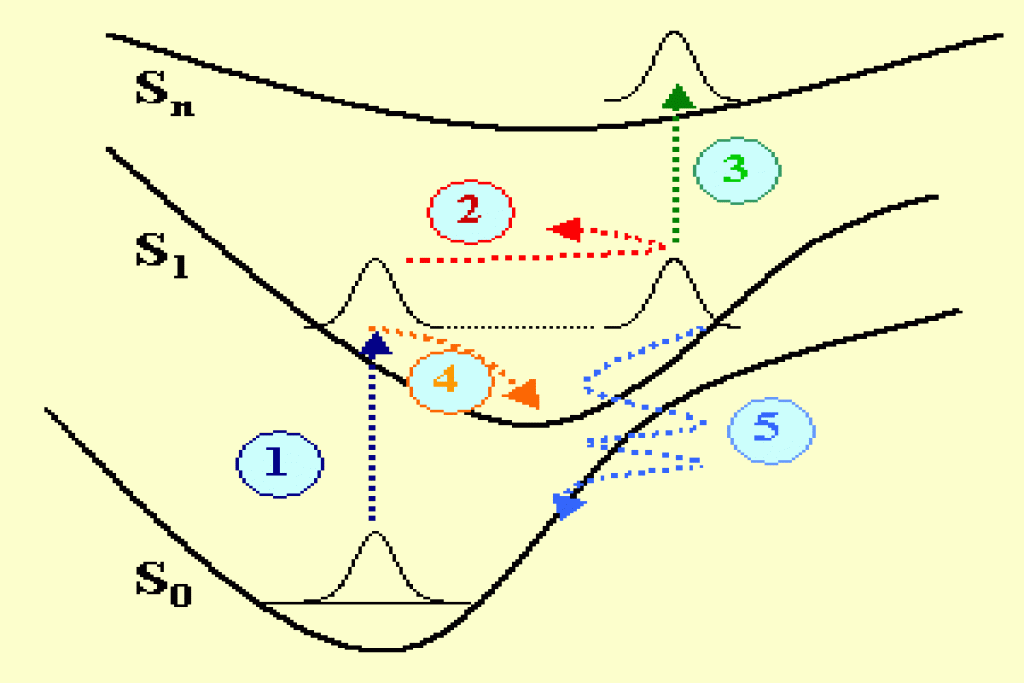
Plusieurs mécanismes interviennent dans l’amortissement du paquet d’ondes. Mentionnons par exemple l’absorption vers les états excités (mécanisme 3 en vert), la relaxation interne vibrationelle (mécanisme 4 en orange) ou encore le transfert tunnel quantique qui se produit dans la région de croisement de potentiel dans l’espace des configurations (mécanisme 5 en bleu clair). D’autre processus (non représentés sur la fig. 1) contribuent également à la relaxation d’énergie, tels que : le couplage spin-orbite entre états singulets et triplets, l’isomérisation, la relaxation radiative vers l’état fondamental (fluorescence).
D2. Dynamique de paquets d’ondes d’une chaîne de polymère
Dans les polymères conjugués, la délocalisation des orbitales moléculaires π, qui est associée aux électrons non hybridés du carbone de leur squelette, est à l’origine de leur grande susceptibilité optique nonlinéaire. De plus, la dynamique moléculaire d’une chaîne de polymère est souvent très influencée par les vibrations d’élongation entre atomes de carbone du squelette. Conceptuellement, cela simplifie l’espace de configuration dynamique en n’utilisant que peu de coordonnées spatiales malgré le fait que, d’un point de vue chimique, le polymère soit un système complexe qui contient plusieurs milliers d’atomes. Les électrons délocalisés sont fortement couplés aux vibrations du squelette, ce qui donne lieu à des états vibroniques bien définis que l’on peut observer dans le spectre d’absorption linéaire du polymère. Un exemple type est celui du polydiacétylène. La figure 2a) montre le squelette du polydiacétylène p-DCH constitué de groupements carbazole latéraux. Dans l’état fondamental de ce polymère, la configuration la plus stable est acétylénique, le squelette étant constitué de liaisons carbone-carbone simples, doubles et triples (fig. 2a). Ces liaisons donnent lieu à deux répliques vibroniques observées du côté haute énergie de la transition optique principale entre l’état fondamental 11Aget l’état excité 11Bu. Cet état excité est souvent appelé exciton X. Il s’agit d’un exciton qui possède un caractère de transfert de charge avec un rayon de Bohr moyen qui s’étend sur quelques monomères. La transition excitonique et les répliques vibroniques sont montrées sur la fig. 2b) dans l’absorption du p-DCH.
Figure 2 : Chaîne de Polydiacétylène p-DCH (2a) et absorption linéaire (2b)
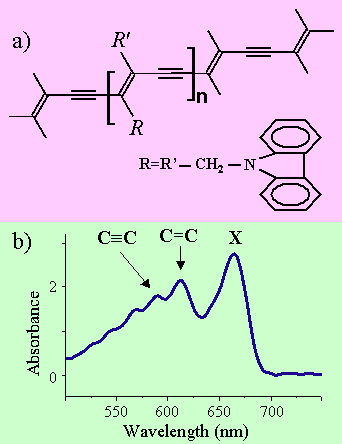
Figure 3 : Dynamique de paquets d’ondes associée à la liaison C=C dans le polydiacétylène p-DCH
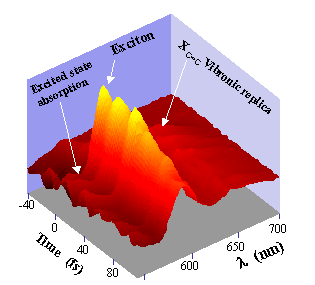
Nous avons étudié la dynamique femtoseconde de films quasi-épitaxiés du polydiacétylène p-DCH déposés sur un substrat d’acide phtalique de potassium. Des mesures pompe-sonde ont été réalisées avec des impulsions de 9 fs de durée obtenues à partir d’un continuum généré dans une fibre optique. Le spectre large de l’impulsion pompe de 9 fs permet d’exciter initialement une superposition cohérente d’états vibroniques. Le paquet d’ondes non stationnaire correspondant est alors sondé à différentes longueurs d’ondes en fonction du retard τ de l’impulsion de sonde de 9 fs. La figure 3 montre la variation spectro-temporelle différentielle. Plusieurs comportements peuvent être distingués. Premièrement, des oscillations très contrastées ayant une période de 23 fs sont observées. Leur contraste est maximum près de la résonance excitonique. Ces oscillations correspondent au paquet d’ondes de la double liaison C=C (23 fs ou 1450 cm-1). Elles sont rapidement amorties avec une constante de temps de 120 fs. De manière complémentaire, un signal oscillatoire négatif se produit vers les grandes longueurs d’ondes. Il est relié à une absorption induite vers un état excité depuis l’état excitonique.
Figure 4 : Dynamique de paquet d’ondes dans les régions spectrales excitonique et d’absorption induite du p-DCH
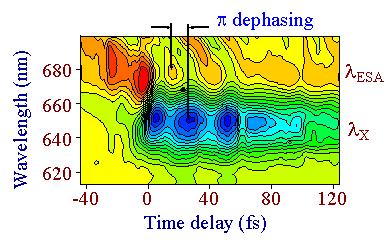
Comme on le voit sur les courbes de niveau de la figure 4, l’analyse détaillée du mouvement du paquet d’ondes montre un déphasage temporel de π entre les oscillations se produisant dans la région spectrale d’absorption (centrée à λESA et celles qui se produisent dans la région excitonique (centrée à λX). ce déphasage de π est dû à l’excursion du paquet d’ondes dans l’espace des configurations comme cela est montré sur la figure 1. Le paquet d’ondes est d’abord engendré dans la région Franck-Condon reliant l’état fondamental 11Ag et l’état excité 11Bu. Il évolue alors dans l’état excité qui possède un décalage Stokes par rapport au niveau fondamental. Après une demi période, il entre dans une région du potentiel où l’absorption induite vers l’état excité supérieur peut se produire. Remarquons que les signaux pompe-sonde ont des signes opposés dans ces deux régions spectrales (signe positif : blanchiment induit ; signe négatif : absorption induite).
D3. Modélisation des états fondamental et excité de polydiacétylènes
Des calculs de chimie quantique des surfaces de potentiel des états fondamental et excité de deux conformations d’isomères PDA (acétylénique) et PBT (butatriénique) ont été faits en utilisant des méthodes de champ auto-cohérent (CASSCF, CASPT2). Les calculs ont été effectués pour le monomère, une chaîne linéaire de dimères et de trimères ainsi que pour une chaîne cyclique de trimères pour chacune des deux conformations. Les principaux résultats sont résumés sur les courbes de potentiels de la figure 5. dans l’état fondamental, la conformation PDA avec la symétrie 11Ag est la plus stable. Contrairement aux calculs Hartree Fock, la conformation PBT ne présente pas d’état stationnaire avec un minimum bien défini de potentiel. Par contre, dans l’état excité, la configuration la plus stable du niveau 21Ag est la PBT qui possède un minimum d’énergie plus bas que l’état 11Bu de conformation PDA. Les potentiels de la figure 5, obtenus pour le modèle du trimère cyclique, permet de comprendre la relaxation ultra-rapide observée dans les polydiacétylènes.
Initialement, lorsque le polymère est excité avec une impulsion laser ultra-brève, la transition Franck Condon se produit entre les états 11Ag et 11Bu associés à la conformation PDA. La relaxation dans l’état excité se produit alors avec un transfert d’énergie entre l’état 11Bu de conformation PDA vers l’état 21Ag de conformation PBT. Puisque ce processus ne nécessite pas de vaincre une barrière de potentiel il est extrêmement rapide. Une gamme de temps typique pour cela est de quelques périodes vibrationnelles des modes les plus actifs de la chaîne (les deux potentiels, qui en théorie sont orthogonaux, doivent être couplés par d’autres degrés de liberté comme les torsions de la chaîne). En ce sens, on peut attribuer l’étape initiale de relaxation, qui a été observée dans plusieurs types de polydiactéylènes en environ 100 fs, à un piégeage de l’exciton dans l’espace des configurations. Ainsi, le potentiel 11Bu de conformation PDA peut être identifié à un exciton libre et le potentiel 21Ag de conformation PBT à un exciton piégé. Nos études expérimentales et théoriques montrent qu’une relaxation géométrique d’une chaîne de polydiacétylène se produit de manière ultra-rapide entre deux conformations différentes de la chaîne.
Figure 5 : Relaxation géométrique ultra-rapide d’une chaîne de polydiacétylène.
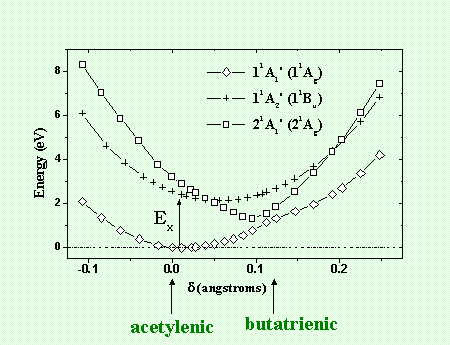
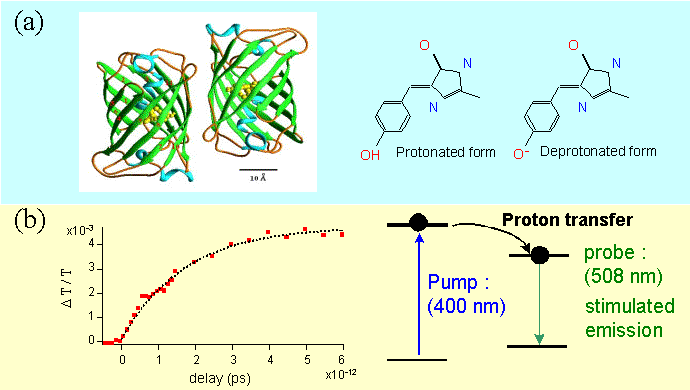
Figure 6 : Structure (a) et mécanisme de transfert de proton dans la protéine fluorescente verte.
D4. Dynamique de transfert de proton dans la protéine fluorescente verte (GFP)
La protéine fluorescente verte est une protéine naturelle (GFPwt), qui a été découverte dans la méduse Aequorea Victoria (structure montrée sur la figure 6a). Après avoir été décodée, la GFP est maintenant couramment utilisée et exprimée dans des organismes vivants et plusieurs variétés mutantes ont été obtenues par ingénierie moléculaire. Ces mutants possèdent des bandes d’émission différentes dans la partie bleu-vert du spectre optique ainsi que des rendements de fluorescence très importants qui les rendent très efficaces comme marqueurs en biologie. Les propriétés photo-physiques de la GFP sont liées à un mécanisme de transfert de proton dans l’état excité. Le transfert de proton se produit en quelques picosecondes et l’état excité dé-protoné se recombine spontanément vers l’état fondamental en émettant des photons. Nous avons étudié la dynamique femtoseconde d’un mutant uv (GFPuv) qui possède un maximum de fluorescence à 508 nm. La dynamique de gain au pic de fluorescence montre que le transfert se produit en 1.7 ps (figure 6b).
D5. De l’étude de la dynamique femtoseconde de la GFP à la sélection d’anticorps dirigés contre le cancer du col de l’utérus
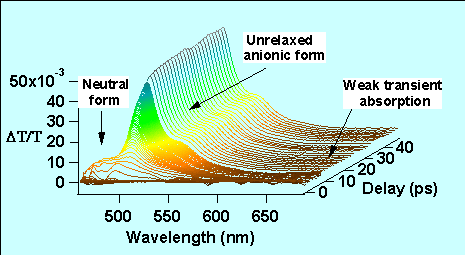
Nous avons étudié en détail la dynamique spectro-temporelle de la GFPuv (figure 7). La dynamique pompe-sonde femtoseconde de gain a été explorée dans la région spectrale de la fluorescence. L’impulsion pompe (400 nm) est obtenue à partir de la génération de deuxième harmonique de la fréquence fondamentale du laser Ti:Saphir et la sonde (480-700 nm) est obtenue à partir de la génération de continuum dans une lame de saphir. Une croissance mono-exponentielle dans la dynamique du gain à 508 nm traduit le fait que le transfert de proton se produit en 1.7 ps (voir également la fig. 6b). D’autres études de la GFP naturelle (GFPwt) ont révélé plusieurs constantes de temps au pic de fluorescence. Ces différences entre la GFPuv et la GFPwt sont attribuées à une meilleure homogénéité des échantillons synthétisés de GFPuv dans lesquels la population de protéines dans la forme anionique est fortement réduite à la température ambiante. De plus, l’étude en solution d’un analogue du chromophore situé dans le cylindre β de la protéine GFP, montre que l’interaction entre la cage protéique et le chromophore joue un rôle déterminant dans la dynamique de la GFP. Cela se manifeste par une absorption induite importante vers les grandes longueurs d’ondes du chromophore synthétique en solution qui, par comparaison, est faible dans le cas de la GFPuv.
Nous avons récemment utilisé la sensibilité à l’environnement du rendement de fluorescence de la GFPuv pour étudier la solubilité de fragments d’anticorps scFv. Les fragments scFv sont des anticorps utiles contre l’oncoprotéine E6 qui joue un rôle actif dans la prolifération des cellules cancéreuses produites par papillomavirus, qui est responsable du cancer du col de l’utérus. Une bonne sensibilité des fragments d’anticorps scFv nécessite qu’ils soient bien repliés. Une fusion covalente de scFv avec la protéine GFPuv permet ainsi d’étudier le repliement de la GFp plutôt que celui de scFv. L’analyse de la dynamique de fluorescence de la GFPuv est donc indirectement un bon reporteur de l’état de repliement de l’anticorps lui même.
Figure 7 : Dynamique spectro-temporelle de la protéine fluorescente verte GFPuv.