D1. Relaxation mechanisms in molecular systems
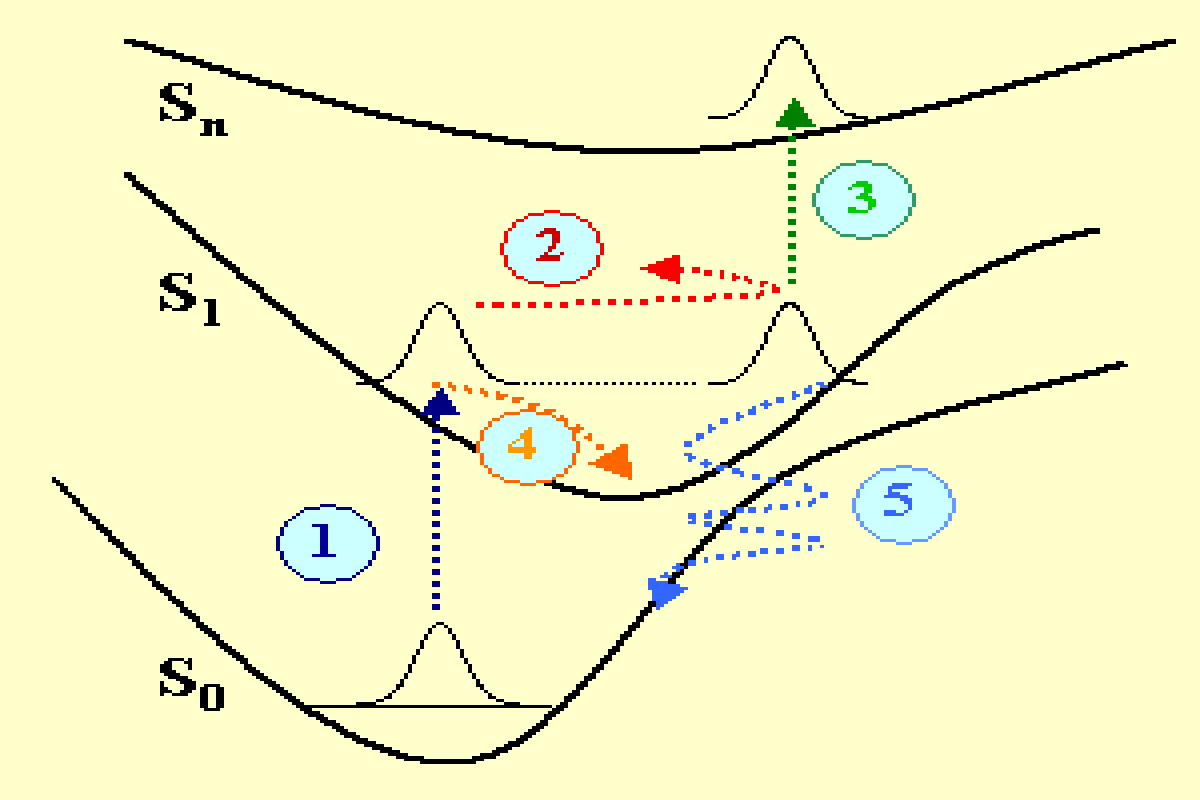
Chemical reactions or photophysical transformations of molecules have their origin in the primary events taking place at the nanometer scale and in the femtosecond temporal scale. These events strongly depend on the molecular dynamics that can be observed using femtosecond optical spectroscopy techniques. In figure 1 several physical mechanisms are represented in a potential in a simplified configuration space. A molecule having three electronic singlet states S0, S1, Sn, is excited with an ultrashort laser pulse with a duration of a few femtoseconds. The initial optical transition from the ground state to S1 (mechanism 1 in dark blue) contributes to excite a coherent superposition of states, i.e. a wave packet that is out of equilibrium and moves forth and back on the excited potential (mechanism 2 in red). A wave packet is also excited in the ground state (not represented). The energy and periodicity of the vibronic wave packets naturally depend on the specific molecular vibrations that are excited (stretching, bending, rotational degrees of freedom of the molecule bonds) as well as on the specific shape of the potential surfaces in the configuration space (Frank-Condon connected potentials, anharmonicity…).
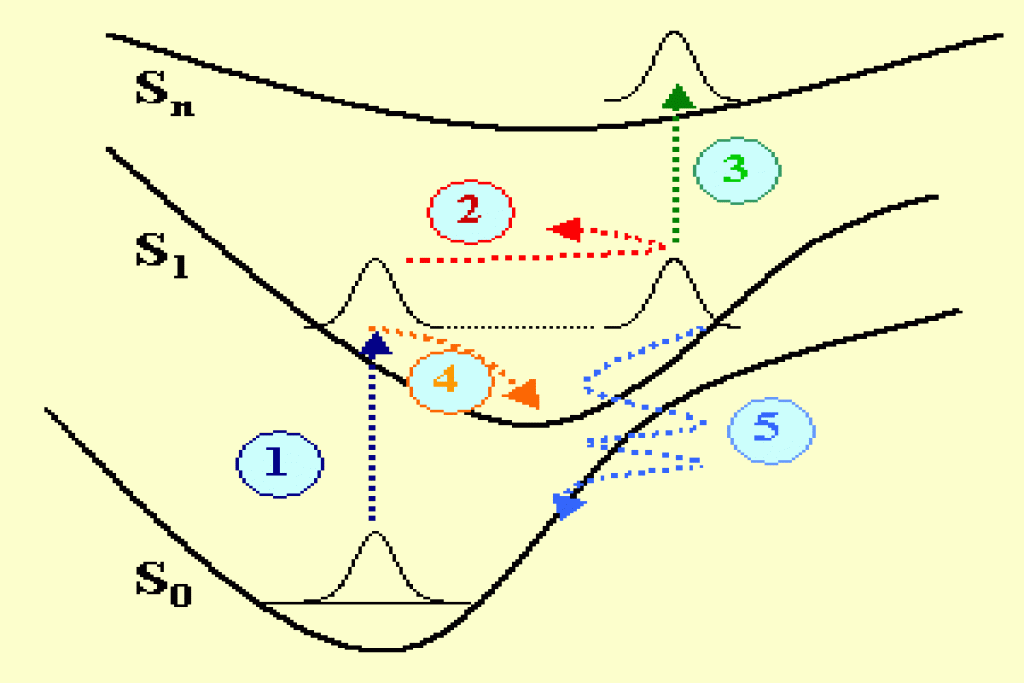
The damping of the wave packets occurs via several additional mechanisms like an excited state absorption (mechanism 3 in green), an internal vibrational relaxation (mechanism 4 in orange) or a quantum tunelling occurring in a region of inter-crossing potentials in the configuration space (mechanism 5 in light blue). There are additional processes (not shown in fig. 1) that contribute to the energy relaxation, like: the singlet-triplet spin orbit coupling, the isomerization, the radiative relaxation to the ground state.
D2. Wave packet dynamics of a polymer backbone
The delocalization of the π molecular orbitals in conjugated polymers, which are associated to the non hybridized carbon electrons of their backbones, is at the origin of their large nonlinear optical susceptibilities . In addition, the molecular dynamics of a polymer chain is often dominated by the carbon-carbon stretching vibrations that occur along the polymer backbone. Conceptually, this simplifies the dynamical configuration space by using a few space coordinates in spite of the fact that, from a chemical point of view, the polymer is a complex system which may contain thousands of atoms. The delocalized electrons are strongly coupled to the vibration manifold of the polymer backbone leading to well defined vibronic states which can be observed in the linear absorption spectrum of the polymer. A typical example is the one of polydiacetylene. Figure 2a) shows the backbone of the polydiacetylene p-DCH constituted of carbazole lateral groups. In the ground state of this polymer, the most stable configuration is the acetylenic one where the backbone consists of single, double and triple carbon-carbon bonds (fig2a). These bonds give rise to two vibronic replica observed on the high energy side of the main optical transition between the 11Ag ground state and the 11Bu excited state. This excited state is often referred to an exciton X with a charge transfer character and which has an average Bohr radius extending over a few monomers distances. The excitonic transition and vibronic replica are displayed in the absorbance of p-DCH shown in fig. 2b).
Figure 2 : Polydiacetylene p-DCH backbone (2a) and linear absorption (2b)
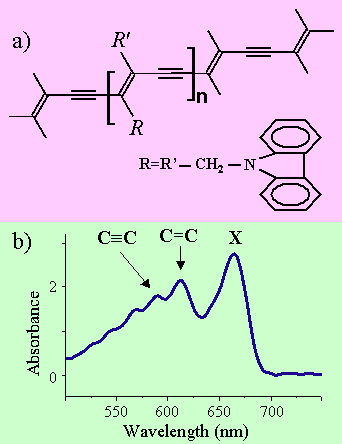
Figure 3 : Wave packet dynamics associated to the C=C stretch in the polydiacetylene p-DCH
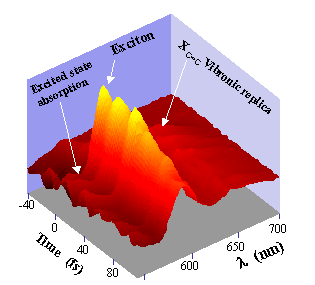
We have investigated the femtosecond dynamics of quasi epitaxial p-DCH polydiacetylene films deposited on a Potassium Acid Phtalate substrate. Pump and probe measurements have been performed using 9 fs laser pulses issued from a broad band continuum generated in an optical fiber. The broad band spectrum of the chirped compensated 9 fs pulses initially sets a coherent superposition of vibronic excited states. The associated non stationary wave packet is then probed at different wavelengths as a function of the pump probe delay τ. Figure 3 shows the spectro-temporal variation of the differential transmission. Several features can be distinguished. First, well contrasted oscillations with a period of 23 fs are observed. Their contrast is maximum near the excitonic resonance. These oscillations correspond to the C=C double bond wave packet (23 fs or 1450 cm-1). They are rapidly damped with a time constant of 120 fs. Correspondingly an oscillatory negative signal occurs on the long wavelength side. It is related to an excited absorption from the excitonic state to higher excited levels.
Figure 4 : Wave packet dynamics in the excitonic and excited state absorption spectral regions of p-DCH
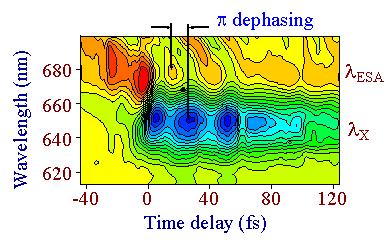
As shown in figure 4, a detailed contour plot analysis of the wave packet motion shows a π temporal dephasing between the oscillations occurring in the excited state absorption spectral region (centered at &lambda,ESA and the ones occurring at the exciton (centered at λX. This π dephasing is due to the excursion of the wave packet in the configuration space as sketched in figure 1. The wave packet is first generated in the Franck Condon region connecting the 11Ag ground state and the 11Bu excited state. The wave packet then evolves in the excited state which is Stokes shifted with respect to the ground state. After half a period, it enters in a favorable potential region so that an excited state absorption takes place. Note that the pump-probe signals have opposite signs for the exciton wave packet (induced bleaching) and the one associated to the excited state region (induced absorption)
D3. Modeling the ground and excited states of polydiacetylenes
Quantum chemical calculations of the ground and excited potential surfaces of two isomer conformations PDA and PBT have been made using methods of complete active space self-consistent field (CASSCF, CASPT2)[9]. The calculations have been performed on the monomer, linear dimer and trimer as well as on the cyclic trimer for each conformation. The main results, are summarized in the potential surfaces of figure 5. In the ground state, the PDA conformation with symmetry 11Ag is the most stable. In contrast to Hartree Fock calculations, the PBT conformation does not exhibit a stationary state with a well defined potential minimum. In the excited state, the most stable configuration is the 21Ag PBT conformation which has an energy minimum lower than the 11Bu PDA conformation. The potentials of figure 5, obtained for the model cyclic trimer representation of the polymer, allows the understanding of the ultrafast relaxation dynamics observed in the polydiacetylenes.
When the polymer is excited with an ultrashort optical pulse, initially the Franck Condon transition occurs between the 11Ag and 11Bu states associated to the PDA conformation. The relaxation on the excited state then takes place via an energy transfer from the 11Bu PDA to the 21Ag PBT conformations. Since this process does not require to overcome a potential barrier, it is very fast. A typical time scale is a few vibrational periods of the most active modes of the chain (the two potentials, which in the theory are orthogonal states, must be coupled via other degrees of freedom like torsions of the chain). In this sense one can attribute the initial step of energy relaxation, which has been observed to occur in several polydiacetylenes within 100 fs, to an exciton trapping in the configuration space. The 11Bu PDA potential can be identified to a free exciton and the 21Ag PBT to a trapped exciton. Our experimental and theoretical studies therefore demonstrate that an ultrafast geometrical relaxation of a polymer chain takes place between two different conformations.
Figure 5 : Ultrafast geometrical relaxation of a polydiacetylene chain.
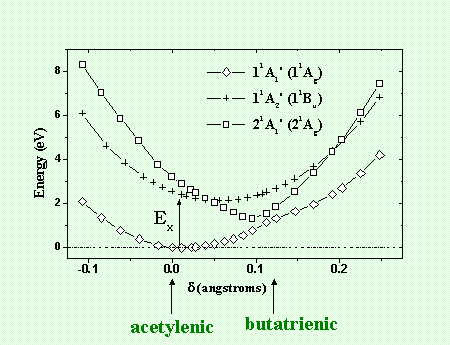
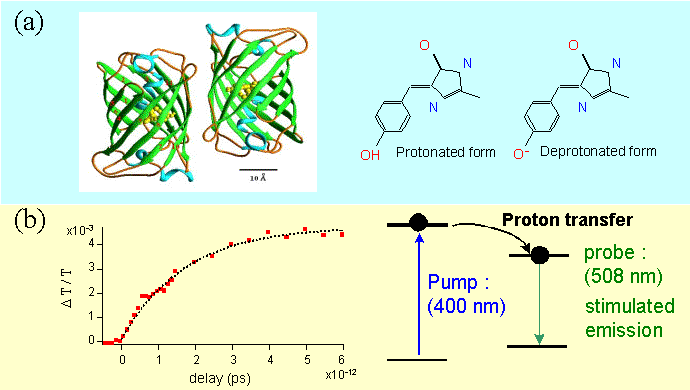
Figure 6 : Structure (a) and proton transfer mechanism in the Green Fluorescent Protein.
D4. Proton transfer dynamics in the Green Fluorescent Protein
The Green Fluorescent Protein is a wild type protein (GFPwt), first discovered in the jelly fish Aequorea Victoria (structure displayed in figure 6a) . After its decoding the GFP is now commonly expressed in living cells and several mutants have been obtained by molecular engineering. These mutants have various emission bands in the blue-green spectrum as well as large fluorescence quantum yields which make them very attractive as efficient fluorescent markers in biology. The photophysical properties of GFP are related to a mechanism of proton transfer in the excited state. The proton transfer occurs in a few picoseconds and the deprotonated excited state spontaneously recombines to the ground state by emitting photons. We have studied the femtosecond dynamics of a uv mutant (GFPuv) with a maximum of fluorescence at 508 nm. The gain dynamics at the fluorescence peak shows that the proton transfer occurs within 1.7 ps (figure 6b).
D5. From the study of the GFP femtosecond dynamics to a selection of antibodies directed against the uterus cancer
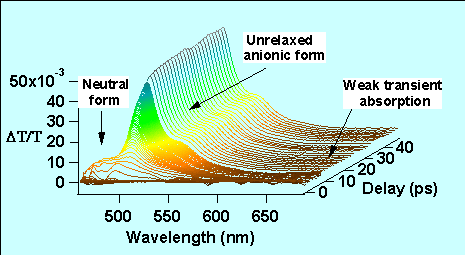
We have studied the detailed spectro temporal dynamics of the GFPuv (figure 7). The femtosecond pump-probe gain dynamics has been investigated in the spectral region of the fluorescence. The pump pulse (400 nm) is obtained from the second harmonic generation of the fundamental 800 nm Ti:Sapphire laser while the probe (480-700 nm) is obtained by a continuum generation in a sapphire plate. A mono-exponential raise of the gain dynamics at 508 nm shows that the proton transfer occurs within 1.7 ps (see also fig. 6b). In contrast other studies of the wild type GFP have revealed several time constants at the fluorescent peak. These differences between the GFPuv and GFPwt are attributed to the improved homogeneity of the GFPuv sample in which the population of the anionic form is strongly reduced at room temperature. In addition, the study in solution of a synthetic analog of the chromophore present inside the β barrel GFP protein shows the importance of the interaction between the proteic cage and the chromophore in the dynamical behavior of GFP. It manifests by a large induced absorption in the long wavelength side of the spectrum of the synthetic soluted chromophore which is very weak in the case of the homogeneous GFPuv
We have recently used the environment sensitivity of the fluorescent yield of GFPuv to study the solubility of scFv antibodies fragments. The scFv fragments are useful antibodies against the protein E6 which has an active role in the proliferation of cancer cells produced by papillomavirus, which is responsible for the uterus cancer. A good solubility of the scFv antibody fragments requires that they are well folded. A covalent fusion of the scFv with the GFPuv protein allows studying the GFPuv folding rather than the scFv. The analysis of the fluorescence dynamics of GFPuv is therefore a good indirect reporter of the folding state of the antibody itself.
Figure 7 : Spectro-temporal dynamics of the Green Fluorescent Protein GFPuv.