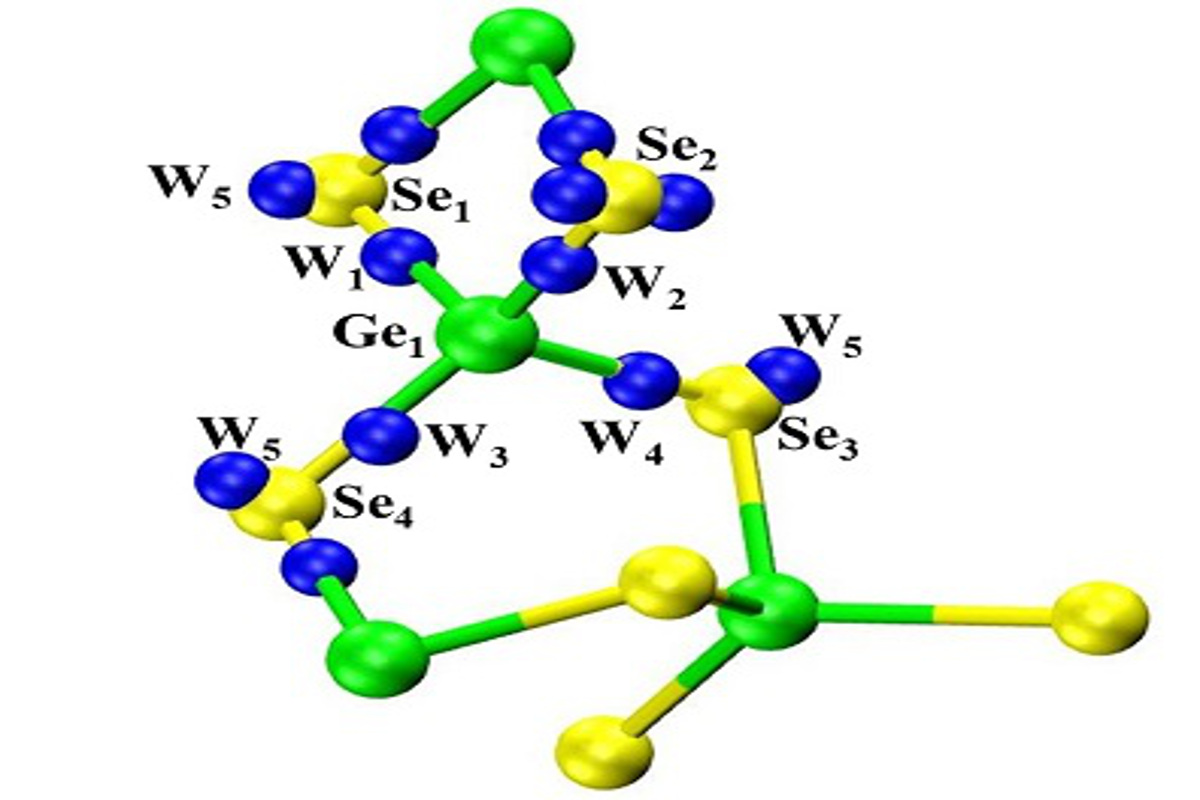
First-principles molecular dynamics is employed to the study of disordered network-forming materials, with a specific emphasis on liquid and glasses of the GexSe(1-x) family. Our approach consists in the production of statistically reliable temporal trajectories from which valuable information can be extracted on structural, vibrational and electronic properties. Historically, involvement in this research line was motivated by the search of the atomic-scale origins for the appearance of the first sharp diffraction peak in the total (and partials) neutron structure factors of liquid GeSe2. This study gave us the opportunity to realise under which conditions the delicate balance between the ionic and the covalent character of bonding (inherent in chalcogenides) can be correctly modeled within density functional theory. In this context, the role of the generalized gradient approximations (GGA) in the expression of the exchange-correlation part of the DFT Kohn-Sham functional has to be underlined. Overall, it appeared that chalcogenides can be taken as a playground having not only very interesting properties to be exploited for applicative purposes, but they can also be considered as crucial systems to benchmark DFT-based models. In AxB(1-x) systems, (A=Ge, Si; B=S,Se,O), one of the most intriguing structural features is the coexistence of a quite regular tetrahedral network with the presence of homopolar Ge-Ge and Se-Se bonds. These latter are a mere consequence of the relative species concentration when x≠ 1/3, since fourfold Ge atoms and twofold Se atoms cannot find their place in tetrahedra on the Ge-rich (x>1/3) and Se-rich side (x>1/3) of the concentration ranges, respectively. However, their occurrence was less expected in the stoichiometric liquid and glassy GeSe2. In agreement with experimental evidence, we were able to rationalize the appearance of homopolar bonds, thereby providing an atomic-scale description of unprecedented quality and setting the scene for a quantitative description of chalcogenides.
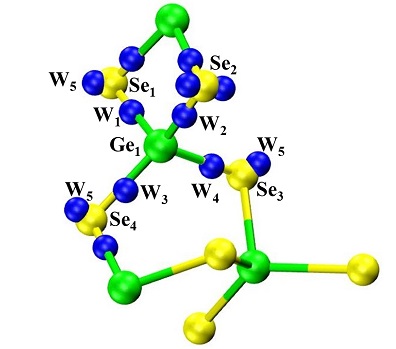
Stimulated by the success on liquid and glassy GeSe2, the availability of a sound theoretical framework paved the way for applications to other systems. Our goal is to attain a predictive power in the production of structural properties such as pair correlation functions, partial structure factors and coordination numbers. Over the years, our approach has been extended to the consideration of different exchange correlation functionals, larger systems (from N=120 to N=480) and a variety of concentrations (GeSe4, Ge2Se3, GeSe), evolving toward the account of pressure effects, very recently addressed in the framework of the PhD thesis of Assil Bouzid.
Recent papers:
- First principles molecular dynamics study of glassy GeS2: atomic structure and bonding properties, M. Celino, S. Le Roux, G. Ori, B. Coasne, A. Bouzid, M. Boero, C. Massobrio, Phys. Rev. B 88, 174201 (2013).
- Compositional threshold and anomalies in connection with stiffness transitions in network glasses, M. Bauchy, M. Micoulaut, M. Boero, C. Massobrio, Phys. Rev. Lett. 110, 165501 (2013).
- The structure of liquid GeSe revisited : a first principles molecular dynamics study, S. Le Roux, A. Bouzid, M. Boero, C. Massobrio, J. Chem. Phys. 138, 174505 (2013).
- Stuctural properties of glassy Ge2Se3 from first-principles molecular dynamics,S. Le Roux, A. Bouzid, M. Boero, C. Massobrio, Phys. Rev. B 86, 224201 (2012).
- Accounting for pressure effects on the calculated equilibrium structure of glassy GeSe2 , A. Bouzid, C. Massobrio, J. Chem. Phys. 137, 046101 (2012).