Nous nous intéressons depuis quelques années à la fonctionnalisation d’un autre type de matériau lamellaire, les oxydes lamellaires, et plus particulièrement d’une phase d’Aurivillius de formule Bi2SrTa2O9 (BST). L’idée générale ici est de pouvoir moduler la ferroelectricité bien connue de cette phase d’Aurivillius[1] pour pouvoir à terme lui conférer des propriétés (ferro)magnétiques par une approche de type « matériaux hybrides ». Mais avant d’arriver aux propriétés, nous nous sommes vite aperçu de la nécessité de développer les outils de fonctionnalisation chimique de ces oxydes lamellaires. Les oxydes sont en général chimiquement et thermiquement plus stables que les hydroxydes que nous étudions par ailleurs. La contrepartie est qu’ils sont également plus difficiles à fonctionnaliser, à cause de la plus forte cohésion du réseau oxygéné.
La fonctionnalisation de ces oxydes lamellaires, par des molécules organiques a été explorée depuis une quarantaine d’année, et connait un regain d’intérêt depuis environ quinze ans. Cependant, la littérature ne fait état que de l’intercalation d’amines et de diamines primaires aliphatiques linéaires. La fonctionalisation par des alcools simples ou des acides phosphoniques a également été décrite. Il n’y a donc aucun résultat concernant l’insertion de molécules « complexes », ne serait-ce que des molécules plus encombrées, ou comportant un groupe aromatique. Par ailleurs et surtout, les réactions d’insertion décrites sont particulièrement longues (les temps de réaction sont typiquement de l’ordre d’une semaine), et donc potentiellement inadaptées à un travail exploratoire sur l’insertion de molécules fonctionnelles.
Il est donc apparu que, pour pouvoir envisager à terme de pouvoir conférer à ces matériaux des propriétés (ferro)magnétiques, il était tout d’abord nécessaire d’en explorer la réactivité plus en détail et d’envisager d’autres voies de fonctionnalisation, permettant d’insérer des molécules plus complexes.
Insertion d’amines
Nous avons donc commencé ce travail en reprenant les réactions décrites dans la littérature, mais en les adaptant afin notamment d’accélérer les temps de réaction. Nous avons pour cela mis au point des protocoles de fonctionnalisation par activation micro-onde. Nous sommes ainsi parvenus à former la phase acide de BST (H2Bi0,1Sr0,85Ta2O7 ou HST) en 3 heures de chauffage micro-onde à 70°C contre 5 jours à température ambiante à pression atmosphérique. Il est à noter que si on effectue la même réaction en chauffant en conditions « classiques », la réaction est incomplète pour le même temps de chauffage, et la cristallinité du composé obtenu bien moins bonne. La fonctionnalisation par des amines et diamines primaires aliphatiques est ensuite effectuée en 2 heures de chauffage micro-onde, à 110°C, contre 7 jours à 66°C (reflux du THF) en conditions classiques. Ainsi, la fonctionnalisation complète (depuis la phase BST) dure 4 heures par activation micro-onde, contre 12 jours en conditions classiques.[2,
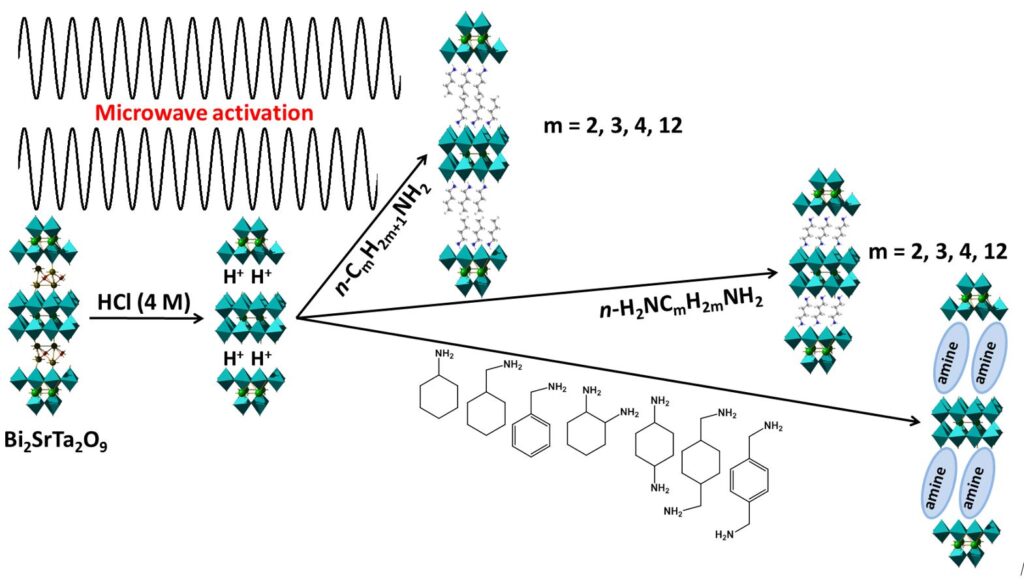
Nous avons ainsi pu optimiser les paramètres réactionnels tels que la nature du solvant ou la quantité d’eau, la température et la durée de la réaction.
Cette augmentation très appréciable de la cinétique de la réaction d’insertion nous a alors permis récemment d’insérer des amines qui n’avaient jamais été insérées jusque-là, telle qu’une amine tertiaire (N,N-diméthyl-hexylamine), ou une amine comportant un groupement aromatique (4-phényl-butylamine). Pour ces réactions, nous avons utilisé un phénomène de « catalyse » par la propylamine ou l’éthylamine : alors que la fonctionnalisation par la N,N-diméthyl-hexylamine ou la 4-phényl-butylamine n’est apparemment pas possible à partir de la phase HST, l’ajout d’une petite quantité de propylamine au milieu réactionnel conduit à la réaction désirée, avec une très bonne cristallinité et sans impureté de C3N-HST.
Il est à noter que parallèlement à ce travail,[3] d’autres résultats similaires utilisant les micro-ondes ont été rapportés dans la littérature, sur la fonctionalisation de phases Dion-Jacobson[4] ou Ruddlesden-Popper.[5]
Insertion-greffage d’alcools
Nous sommes également parvenus à greffer un certain nombre d’alcools et de diols, de manière particulièrement rapide et efficace et nous avons étudié plus particulièrement la réactivité comparée alcools/amines.[6]
Nous avons tout d’abord montré que le greffage d’alcools aliphatiques simples mais aussi aromatiques ou plus encombrés était réalisable avec des procédures sensiblement analogues à celles utilisées pour l’insertion d’amines aliphatiques, en utilisant une stratégie de préinsertion (fonctionnalisation préalable de HST par une amine relativement courte, éthylamine ou butylamine, puis greffage de l’alcool désiré. Néanmoins, nous avons montré que le tert-butanol, très encombré, ne s’insérait qu’à des taux assez faibles.
Nous avons ensuite étudié le greffage de (a,w)-diols, toujours par réactions assistées par micro-ondes. Nous avons montré que pour les diols à chaînes longues (4 atomes de carbone ou plus), les deux fonctions alcool se greffaient à la couche inorganique, mais que pour les alcools plus courts, éthylène glycol et propylène glycol, une seule des deux fonctions se greffait, conduisant à un arrangement en double couche, avec une fonction alcool libre dans l’espace interlamellaire (collaboration avec Fabrice Leroux de l’ICCF à Clermont-Ferrand pour les études en RMN du solide).
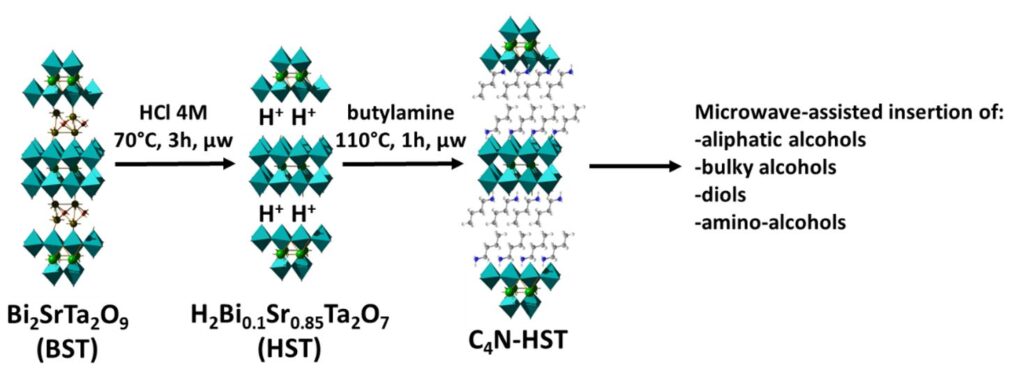
Enfin, pour comparer la réactivité des amines et celle des alcools, nous avons cherché à greffer un (a,w)-amino-alcool, le 5-aminopentan-1-ol. Nous avons montré que l’amine réagissait toujours préférentiellement à l’alcool, mais qu’il était possible de contrôler le greffage ou non de l’alcool en jouant sur la quantité d’eau dans le mélange réactionnel : pour les petites quantités d’eau les deux groupements, amine et alcool, réagissent, conduisant à un mode d’insertion en « pilier » alors que pour les grandes quantités d’eau, seule la partie amine se lie à la couche inorganique, la partie alcool restant libre dans l’espace interlamellaire.
Nous avons enfin montré qu’il était possible de passer de la situation « double couche » à la situation « pilier » par déshydratation progressive. Malheureusement, nous ne sommes pas encore parvenus à réaliser l’évolution inverse, qui est probablement pH dépendante.
Comparaison micro-ondes / réactions solvothermales classiques
Depuis le début de ce travail sur l’insertion dans les oxydes lamellaires, nous avons montré que les réactions solvothermales assistées par micro-ondes étaient particulièrement efficaces, beaucoup plus rapides que les réactions effectuées classiquement (température ambiante ou chauffage classique à pression atmosphérique) et conduisant à des produits avec de très bonnes cristallinités. Ces avantages ont permis d’insérer de nombreuses molécules, de plus en plus complexes. La justification généralement avancée par nous et d’autres pour expliquer cela est que le chauffage micro-onde permet un échauffement rapide et direct du volume réactionnel, sans nécessiter un échauffement long d’un corps métallique comme dans le cas d’un chauffage solvothermal classique, ce qui limiterait les risques de piéger des sous-produits dans un puits de potentiel de la surface réactionnelle.[6–10]
Un peu par hasard, nous avons cependant mis en évidence récemment que la situation était parfois un peu plus complexe, les réactions d’insertion par voie solvothermale sous activation micro-ondes pouvant en fait conduire à d’autres produits que les mêmes réactions conduites par voie solvothermales classiques.[11] Ainsi l’insertion de phenylbutylamine, au micro-ondes, pendant 1,5 h conduit à un composé « cinétique » avec une distance interlamellaire de 2,2 nm, tandis que la même réaction, par voie solvothermale classique pendant 18 h, conduit au produit « thermodynamique » avec une distance interlamellaire de 3,0 nm. L’analyse de profils de densité électronique selon la direction perpendiculaire aux plans des feuillets (axe c) montre que les ligands sont largement interdigités dans le cas du composé « micro-ondes », arrangement probablement stabilisé par p-stacking, alors qu’ils forment une double couche sans interdigitation dans le cas du composé « solvothermal ».
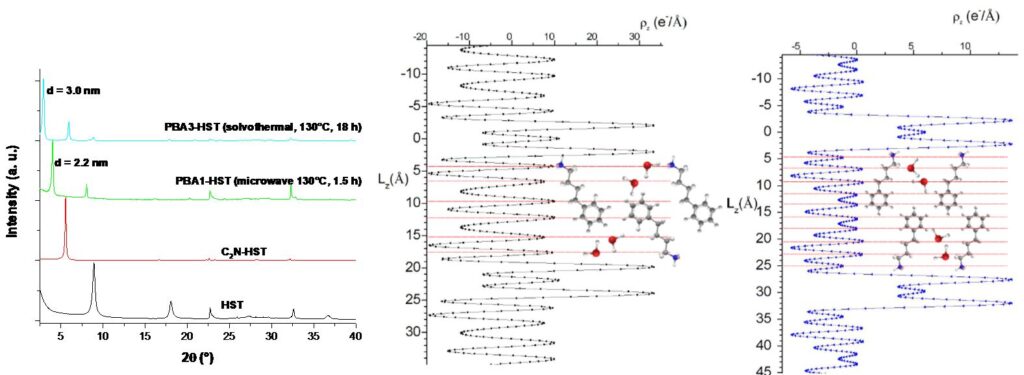
A gauche, diagrammes RX des composés hybrides fonctionnalisés par le phénylbutylamine (PBA) pendant 1,5 h ou pendant 18 h (Cu Kα1 = 0,1540598 nm), au milieu, profil de densité électronique 1D et affinement pour le composé « 1,5 h » et à droite, profil de densité électronique 1D et affinement pour le composé « 18 h » (Collaboration Christine Taviot-Guého, ICCF, Clermont-Ferrand).[11]
Cette étude montre l’activation micro-ondes ne fait pas qu’accélérer les réactions, que les mécanismes et paramètres de réaction sont probablement très complexes, et qu’il reste encore beaucoup de points à explorer pour tirer pleinement profit des réactions d’insertion sous chauffage micro-onde.
De l’insertion à la modification post-synthèse : chimie « click » in situ
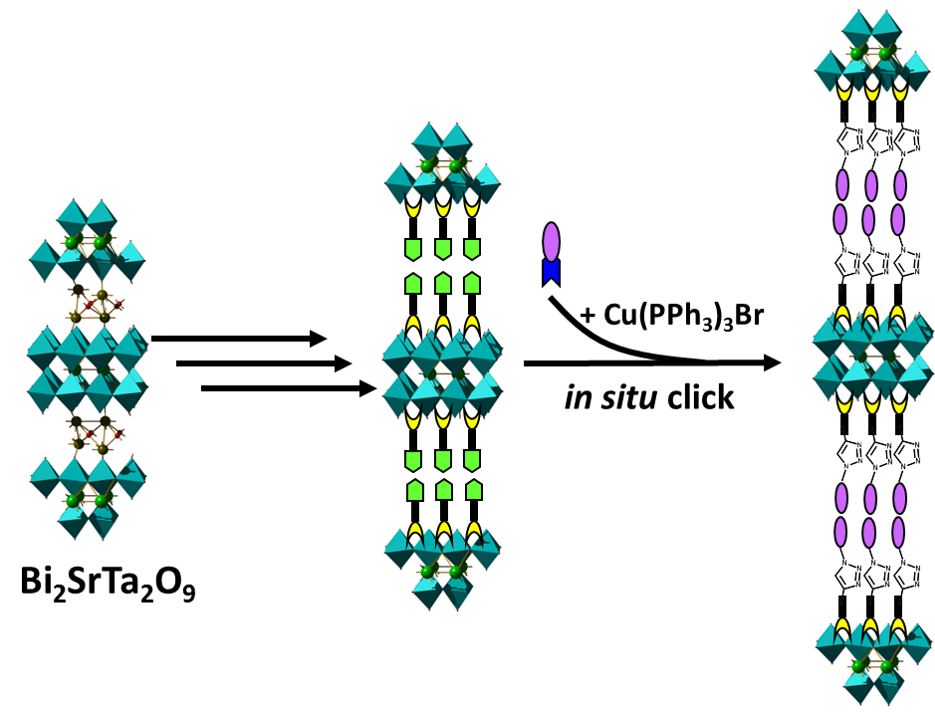
Le greffage d’alcools nous a également permis de mettre au point une nouvelle stratégie pour la fonctionnalisation des oxydes, la synthèse in situ. Cette stratégie est similaire à celle que nous avions décrite pour des hydroxydes simples lamellaires.[12] Ici cependant, les oxydes sont beaucoup plus stables que les hydroxydes, et il est donc possible d’utiliser un plus grand nombre de réactions de chimie organique in situ. Nous avons utilisé les réactions de chimie « click » par voie micro-onde, entre un azide aliphatique en solution, et un alcyne vrai greffé dans l’espace interlamellaire via une fonction alcool primaire. Les résultats montrent une évolution de la distance interlamellaire et une signature infrarouge en accord avec la formation de la molécule. La caractérisation par RMN du solide (collaboration Fabrice Leroux, Clermont-Ferrand) a permis de prouver l’efficacité de la réaction in situ.[13]
[1] E. C. Subbarao, Journal of Physics and Chemistry of Solids 1962, 23, 665–676.
[2] Y. Wang, Hybridation of Layered Oxides : From Insertion to in Situ Synthesis, Université de Strasbourg, 2016.
[3] Y. Wang, E. Delahaye, C. Leuvrey, F. Leroux, P. Rabu, G. Rogez, Inorg. Chem. 2016, 55, 4039–4046.
[4] J. R. Boykin, L. J. Smith, Inorg. Chem. 2015, 54, 4177–4179.
[5] S. Akbarian-Tefaghi, E. Teixeira Veiga, G. Amand, J. B. Wiley, Inorg. Chem. 2016, 55, 1604–1612.
[6] Y. Wang, M. Nikolopoulou, E. Delahaye, C. Leuvrey, F. Leroux, P. Rabu, G. Rogez, Chem. Sci. 2018, 9, 7104–7114.
[7] Y. Wang, E. Delahaye, C. Leuvrey, F. Leroux, P. Rabu, G. Rogez, Inorg. Chem. 2016, 55, 4039–4046.
[8] S. Akbarian-Tefaghi, E. Teixeira Veiga, G. Amand, J. B. Wiley, Inorg. Chem. 2016, 55, 1604–1612.
[9] S. Akbarian-Tefaghi, J. B. Wiley, Dalton Trans. 2018, 47, 2917–2924.
[10] H. J. Kitchen, S. R. Vallance, J. L. Kennedy, N. Tapia-Ruiz, L. Carassiti, A. Harrison, A. G. Whittaker, T. D. Drysdale, S. W. Kingman, D. H. Gregory, Chem. Rev. 2014, 114, 1170–1206.
[11] Y. Wang, C. Leuvrey, E. Delahaye, F. Leroux, P. Rabu, C. Taviot-Guého, G. Rogez, J. Solid State Chem. 2019, 269, 532–539.
[12] O. Palamarciuc, E. Delahaye, P. Rabu, G. Rogez, New J. Chem. 2014, 38, 2016–2023.
[13] Y. Wang, E. Delahaye, C. Leuvrey, F. Leroux, P. Rabu, G. Rogez, Inorg. Chem. 2016, 55, 9790–9797.