We have been interested for some years in the functionalization of another type of lamellar material, lamellar oxides, and more particularly of an Aurivillius phase of formula Bi2SrTa2O9 (BST). The general idea here is to be able to modulate the well known ferroelectricity of this Aurivillius phase[1] in order to be able to confer (ferro)magnetic properties to it by a “hybrid materials” approach. But before reaching the properties, we quickly realized the necessity to develop tools for the chemical functionalization of these lamellar oxides. The layered oxides are in general chemically and thermally more stable than the layered hydroxides that we also study. The counterpart is that they are also more difficult to functionalize, because of the stronger cohesion of the oxygenated network.
The functionalization of these lamellar oxides by organic molecules has been explored for about forty years, and has received renewed interest for about fifteen years. However, the literature only reports the intercalation of amines and linear aliphatic primary diamines. Functionalization with simple alcohols or phosphonic acids has also been described. There are therefore no results concerning the insertion of “complex” molecules, even only bulkier or aromatic ones. Moreover, and above all, the insertion reactions described are particularly long (reaction times are typically of the order of a week), and therefore potentially unsuitable for exploratory work on the insertion of functional molecules.
It thus appeared that, in order to be able to consider eventually conferring (ferro)magnetic properties to these materials, it was first necessary to explore their reactivity in more detail and to consider other functionalization routes, allowing the insertion of more complex molecules.
Insertion of amines
We started this work by using the reactions described in the literature, but by adapting them in order to accelerate the reaction times. We have developed protocols for functionalization by microwave activation. We succeeded in forming the acidic phase of BST (H2Bi0.1Sr0.85Ta2O7 or HST) in 3 hours of microwave heating at 70°C compared to 5 days at room temperature at atmospheric pressure. It is to be noted that if the same reaction is carried out by heating under “classical” conditions, the reaction is incomplete for the same heating time, and the crystallinity of the compound obtained is much less good. The functionalization with primary aliphatic amines and diamines is then performed in 2 hours of microwave heating, at 110°C, against 7 days at 66°C (reflux of THF) under conventional conditions. Thus, complete functionalization (from the BST phase) takes 4 hours by microwave activation, compared to 12 days under conventional conditions.[2,3]
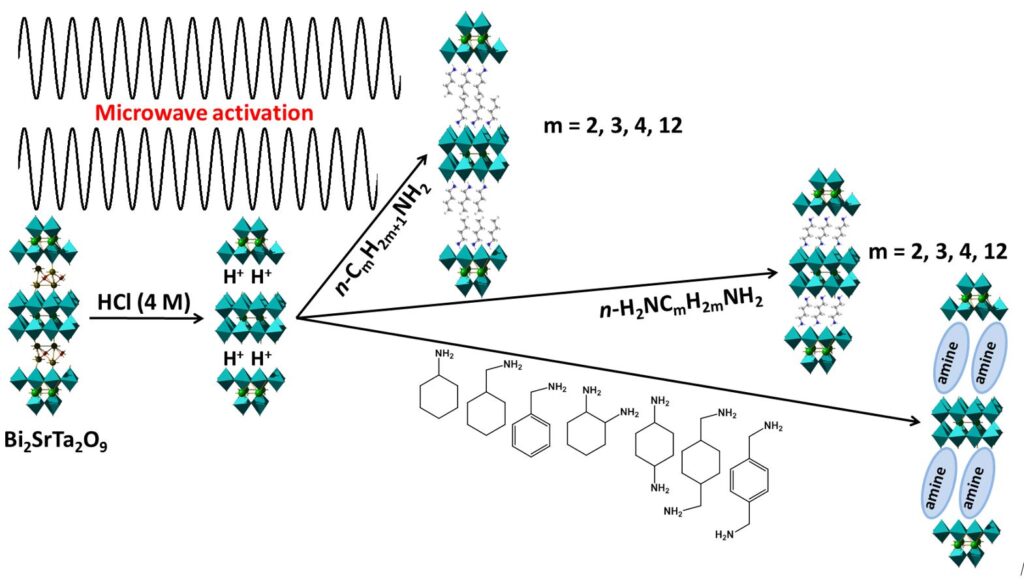
We were thus able to optimize the reaction parameters such as the nature of the solvent or the amount of water, the temperature and the duration of the reaction.
This very appreciable increase of the kinetics of the insertion reaction allowed us to insert amines that had never been inserted before, such as a tertiary amine (N,N-dimethyl-hexylamine), or an amine with an aromatic group (4-phenyl-butylamine). For these reactions, we used a propylamine or ethylamine “catalysis” phenomenon: while functionalization with N,N-dimethyl-hexylamine or 4-phenyl-butylamine is apparently not possible from the HST phase, the addition of a small amount of propylamine to the reaction medium leads to the desired reaction, with very good crystallinity and without C3N-HST impurities.
It should be noted that in parallel to this work,[3] other similar results using microwaves have been reported in the literature, on Dion-Jacobson[4] or Ruddlesden-Popper phase functionalization.[5]
Insertion-grafting of alcohols
We have also managed to graft a number of alcohols and diols, in a particularly fast and efficient way, and we have studied more particularly the compared reactivity between alcohols and amines.[6]
We first showed that the grafting of simple aliphatic alcohols but also aromatic or more bulky alcohols was achievable with procedures substantially analogous to those used for the insertion of aliphatic amines, using a pre-insertion strategy (prior functionalization of HST with a relatively short amine, ethylamine or butylamine, and then grafting of the desired alcohol. Nevertheless, we have shown that tert-butanol, which is very bulky, was inserted only at quite low rates.
We then studied the grafting of (a,w)-diols, again by microwave assisted reactions. We showed that for long chain diols (4 or more carbon atoms), both alcohol functions grafted to the inorganic layer, but for shorter alcohols, ethylene glycol and propylene glycol, only one of the two functions grafted, leading to a double layer arrangement, with a free alcohol function in the interlamellar space (collaboration with Fabrice Leroux from ICCF in Clermont-Ferrand for the solid state NMR studies)
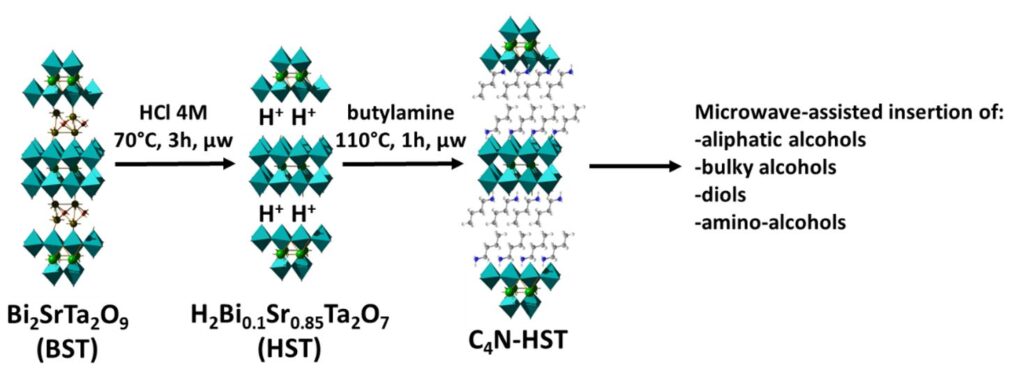
Finally, in order to compare the reactivity of amines and alcohols, we tried to graft an (a,w)-amino-alcohol, 5-aminopentan-1-ol. We showed that the amine always reacted preferentially to the alcohol, but that it was possible to control the grafting or not of the alcohol by playing on the amount of water in the reaction mixture: for small amounts of water both groups, amine and alcohol, react, leading to a “pillar” mode of insertion, whereas for large amounts of water, only the amine part binds to the inorganic layer, the alcohol part remaining free in the interlamellar space.
We have finally shown that it is possible to switch from the “double layer” situation to the “pillar” situation by progressive dehydration. Unfortunately, we have not yet succeeded in realizing the reverse evolution, which is probably pH dependent.
Comparison microwave / classical solvothermal reactions
Since the beginning of this work on insertion into lamellar oxides, we have shown that microwave-assisted solvothermal reactions were particularly efficient, much faster than reactions performed conventionally (room temperature or conventional heating at atmospheric pressure) and leading to products with very good crystallinity. These advantages allowed the insertion of many molecules, more and more complex. The rationale generally advanced by us and others to explain this is that microwave heating allows rapid and direct heating of the reaction volume, without requiring long heating of a metal body as in the case of conventional solvothermal heating, which would limit the risks of trapping byproducts in a potential well on the reaction surface.[6-10]
Somewhat coincidentally, however, we have recently highlighted that the situation is sometimes a bit more complex, as insertion reactions by solvothermal heating under microwave activation can actually lead to other products than the same reactions conducted by conventional solvothermal heating.[11] Thus the insertion of phenylbutylamine, by microwave, for 1.5 h leads to a “kinetic” compound with an interlamellar distance of 2.2 nm, while the same reaction, by conventional solvothermal route for 18 h, leads to the “thermodynamic” product with an interlamellar distance of 3.0 nm. The analysis of electron density profiles along the direction perpendicular to the planes of the sheets (c-axis) shows that the ligands are largely interdigitated in the case of the “microwave” compound, an arrangement probably stabilized by pi-stacking, while they form a double layer without interdigitation in the case of the “solvothermal” compound.
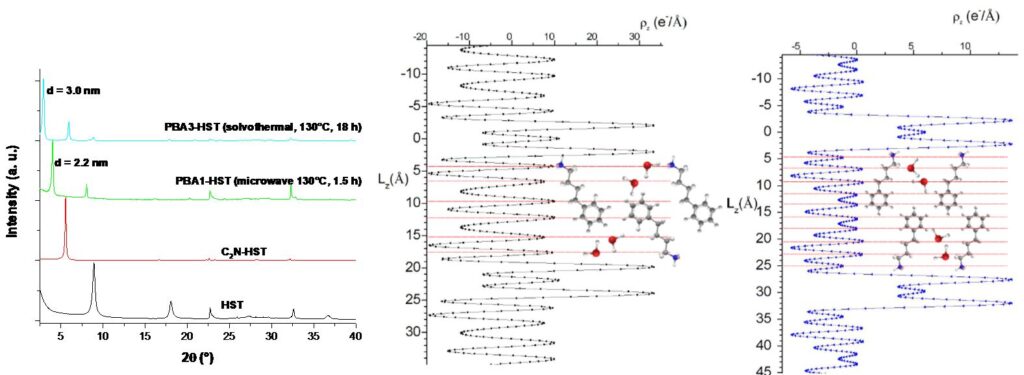
Left, X-ray diagrams of hybrid compounds functionalized by phenylbutylamine (PBA) during 1.5 h or during 18 h (Cu Kα1 = 0,1540598 nm), middle, 1D electron density profile and refinement for the « 1.5 h compound » and right, 1D electron density profile and refinement for the « 18 h compound » (Collaboration Christine Taviot-Guého, ICCF, Clermont-Ferrand).[11]
This study shows that microwave activation does not only accelerate reactions, that the mechanisms and reaction parameters are probably very complex, and that there are still many points to be explored to take full advantage of insertion reactions under microwave heating.
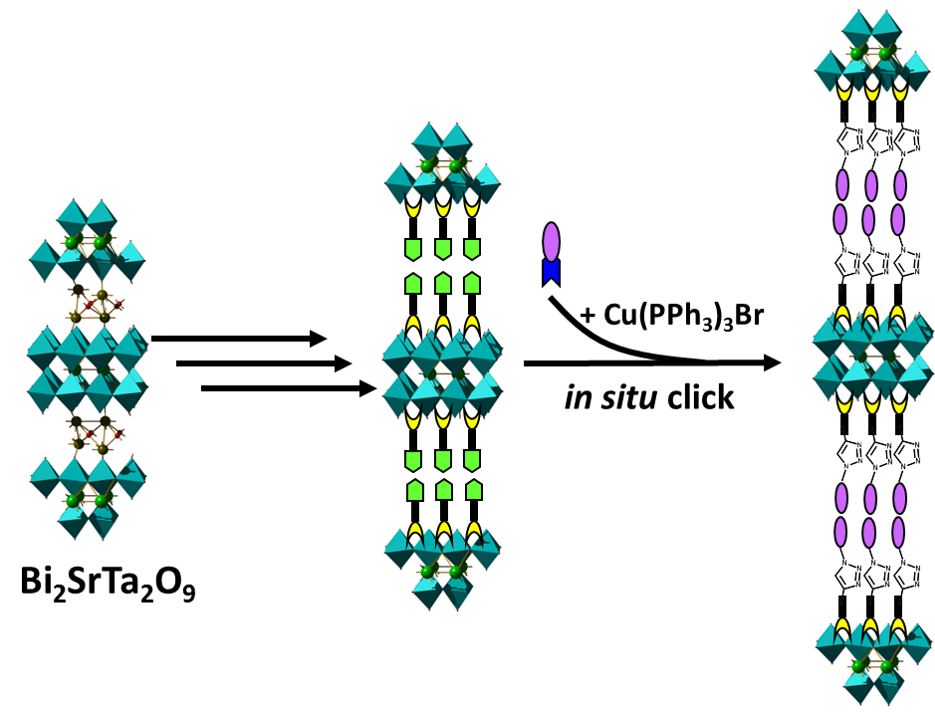
From insertion to post-synthesis modification: in situ “click” chemistry
The grafting of alcohols has also allowed us to develop a new strategy for the functionalization of oxides, the in situ synthesis. This strategy is similar to the one we described for layered simple hydroxides.[12] Here, however, oxides are much more stable than hydroxides, and it is therefore possible to use a larger number of in situ organic chemistry reactions. We have used microwave click chemistry reactions between an aliphatic azide in solution and a true alkyne grafted into the interlamellar space via a primary alcohol function. The results show an evolution of the interlamellar distance and an infrared signature in agreement with the formation of the molecule. The characterization by solid state NMR (Fabrice Leroux collaboration, Clermont-Ferrand) allowed to prove the efficiency of the in situ reaction.
[1] E. C. Subbarao, Journal of Physics and Chemistry of Solids 1962, 23, 665–676.
[2] Y. Wang, Hybridation of Layered Oxides : From Insertion to in Situ Synthesis, Université de Strasbourg, 2016.
[3] Y. Wang, E. Delahaye, C. Leuvrey, F. Leroux, P. Rabu, G. Rogez, Inorg. Chem. 2016, 55, 4039–4046.
[4] J. R. Boykin, L. J. Smith, Inorg. Chem. 2015, 54, 4177–4179.
[5] S. Akbarian-Tefaghi, E. Teixeira Veiga, G. Amand, J. B. Wiley, Inorg. Chem. 2016, 55, 1604–1612.
[6] Y. Wang, M. Nikolopoulou, E. Delahaye, C. Leuvrey, F. Leroux, P. Rabu, G. Rogez, Chem. Sci. 2018, 9, 7104–7114.
[7] Y. Wang, E. Delahaye, C. Leuvrey, F. Leroux, P. Rabu, G. Rogez, Inorg. Chem. 2016, 55, 4039–4046.
[8] S. Akbarian-Tefaghi, E. Teixeira Veiga, G. Amand, J. B. Wiley, Inorg. Chem. 2016, 55, 1604–1612.
[9] S. Akbarian-Tefaghi, J. B. Wiley, Dalton Trans. 2018, 47, 2917–2924.
[10] H. J. Kitchen, S. R. Vallance, J. L. Kennedy, N. Tapia-Ruiz, L. Carassiti, A. Harrison, A. G. Whittaker, T. D. Drysdale, S. W. Kingman, D. H. Gregory, Chem. Rev. 2014, 114, 1170–1206.
[11] Y. Wang, C. Leuvrey, E. Delahaye, F. Leroux, P. Rabu, C. Taviot-Guého, G. Rogez, J. Solid State Chem. 2019, 269, 532–539.
[12] O. Palamarciuc, E. Delahaye, P. Rabu, G. Rogez, New J. Chem. 2014, 38, 2016–2023.
[13] Y. Wang, E. Delahaye, C. Leuvrey, F. Leroux, P. Rabu, G. Rogez, Inorg. Chem. 2016, 55, 9790–9797.