The common aim of the team « Modelling » of the DSI is to provide atomic or electronic level insights into a wide range of materials, in isolated, complexes or condensed phases. DSI’s computational team mainly focuses on the study of supported nanostructures, metals or molecules. Both substrate nanostructuration and nanostructure growth processes are investigated as much as possible in connection with the potential properties (transport, magnetism, reactivity, optics). This involves developing a computational chemistry and physical methodology and expanding the scale and efficiency to provide realistic simulations. Its skills extend therefore from first-principle methods to parametrized methods and classical atomistic numerical simulations. The knowledge of such a range of methodologies allow to treat various materials and phenomena like: surface atomic diffusion, growth processes, ordering in alloys and nanoalloys, adsorption of molecules, magnetism in systems of reduced dimensionality and transport properties in nanojunctions.
An important part of the work of the DSI’s computational team is also the calculation of properties directly comparable with experiment measurements such as X-ray absorption spectrum, X-ray photoemission spectroscopy and STM images.
Research Activities
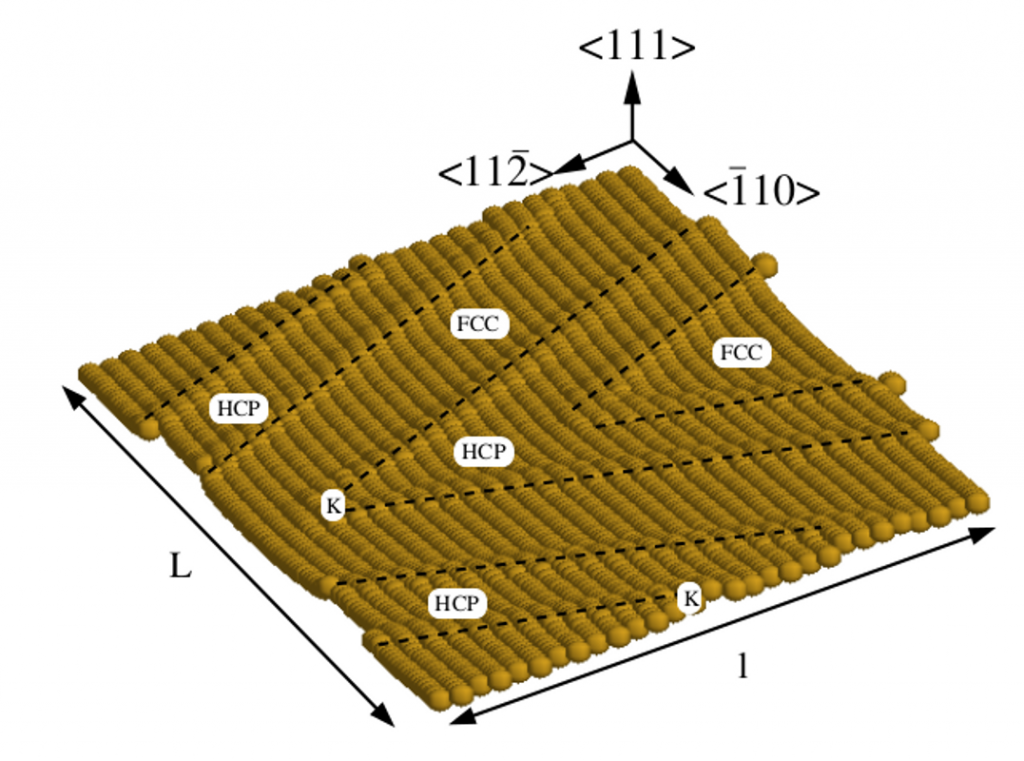

Code Development
Methods
Classical Molecular Dynamics
Monte Carlo Simulations
Electronic Structure Calculations





Recent publications :
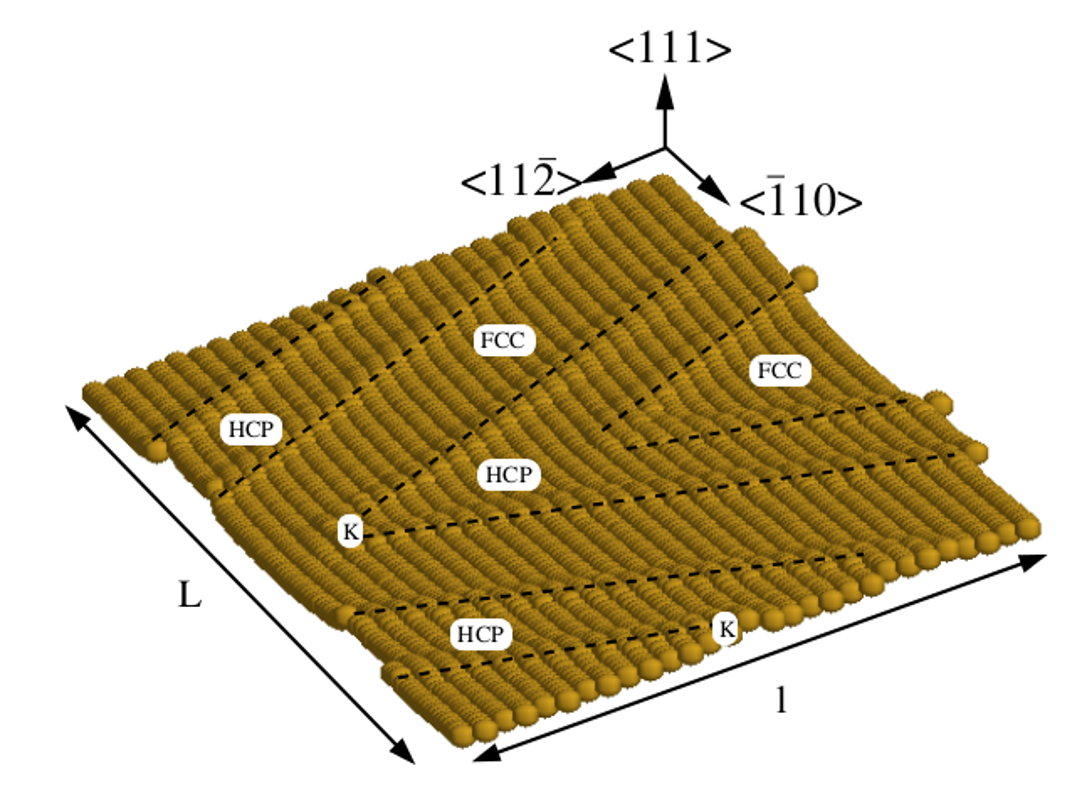
Diffusion, Nucleation & Growth

Surface & Adsorbate Nanostructuration