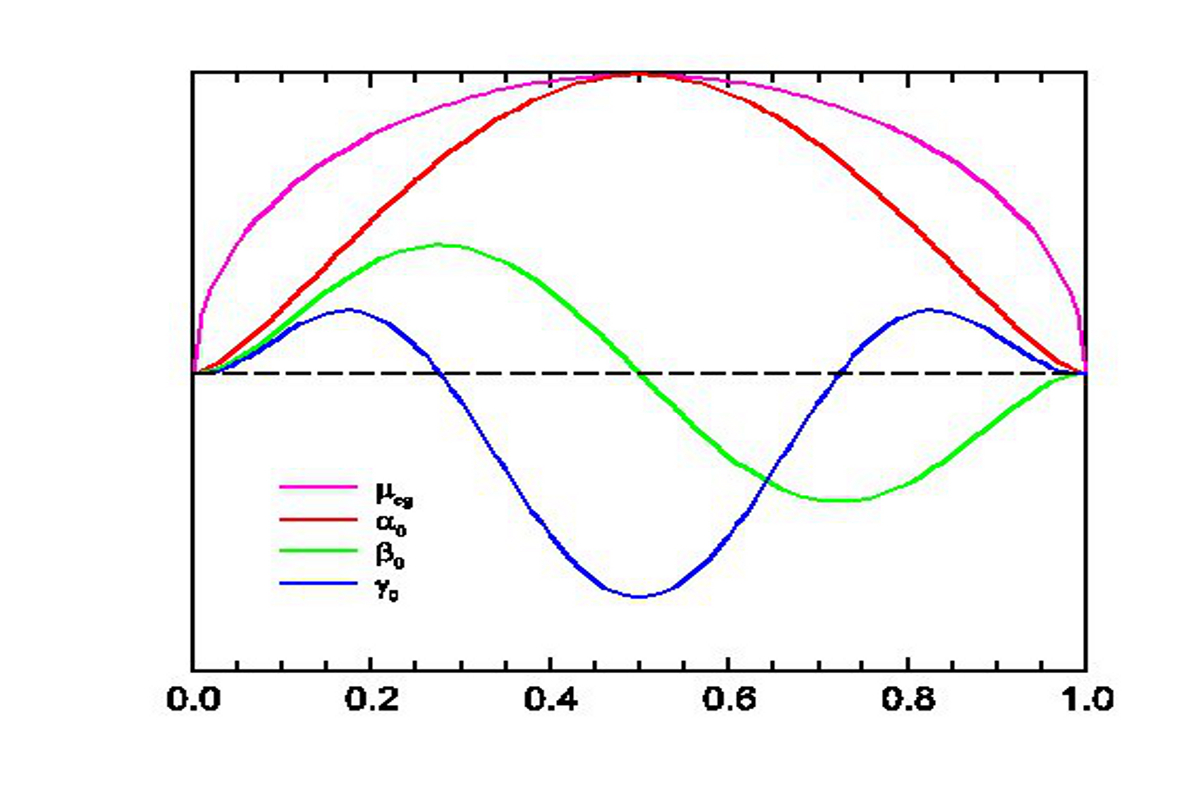
Push-pull molecules made of electron donor and acceptor end groups connected via a conjugated backbone are indispensable to functionalize polymeric materials in optoelectronic applications because they combine the ability to be oriented in electric fields with quadratic non linear optical (NLO) responses. The prospects for linear compounds with a pronounced charge transfer character should be especially promising since they possess large ground state dipole moments, polarizability anisotropies, and negative quadratic hyperpolarizabilities. Indeed, our present understanding of NLO responses in charge transfer compounds can be rationalized by a set of universal response functions versus the ground state ionicity. These have largely been tested experimentally by piece-wise assembling measurements performed in solvents of increasing polarity on series of chromophores with varying acceptor and donor groups and/or conjugation paths.
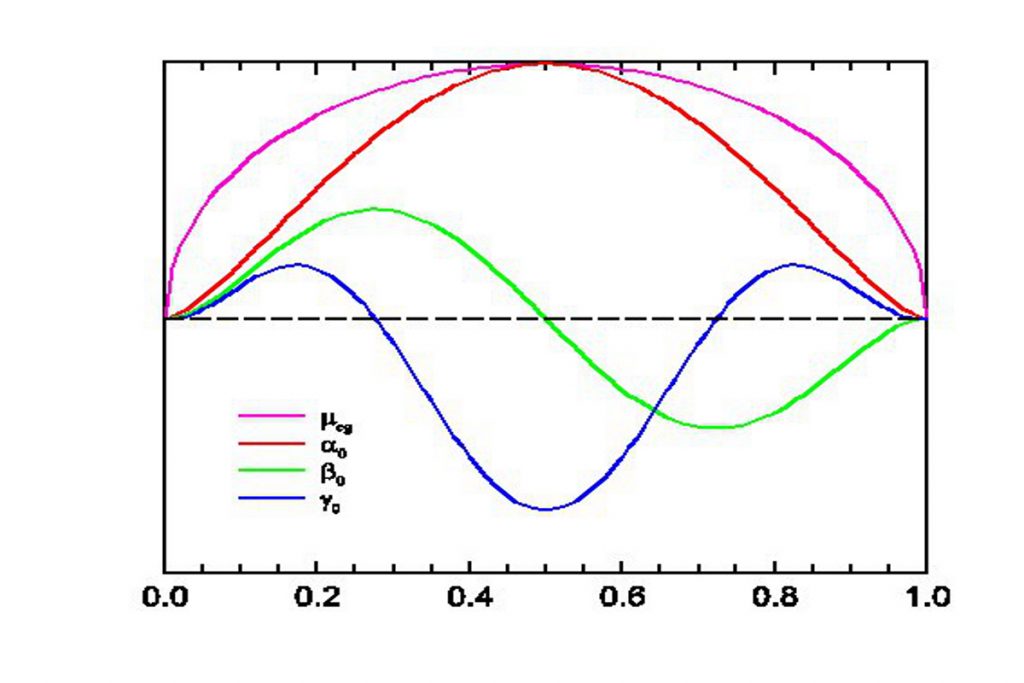
In para-pyridinium-phenoxides, a class of biaryls, the resonance energy of aromatic rings favors a twisted, benzenoid, charge transfer configuration over the planar, quinonoid, neutral form.
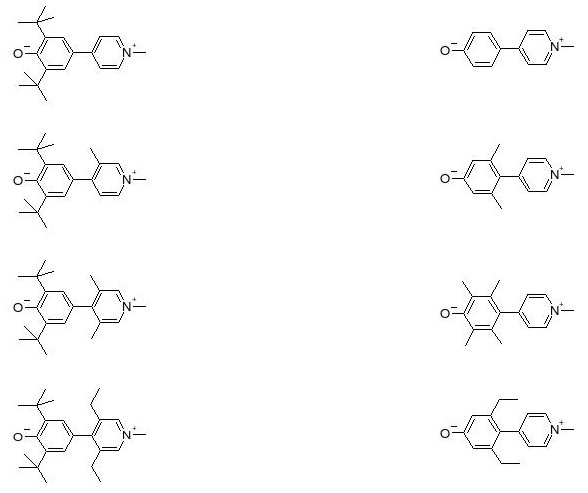
The sensitivity of these compounds to the dielectric and chemical natures of their environments designates them as good candidates for checking the validity of formalisms relating molecular structure and NLO properties. This opens up a great opportunity to investigate relationships between steric and electronic interactions on the responses of zwitterionic chromophores and to test the numerical predictions at the semi-empirical, density functional, and ab initio levels of theory for various solvation models.
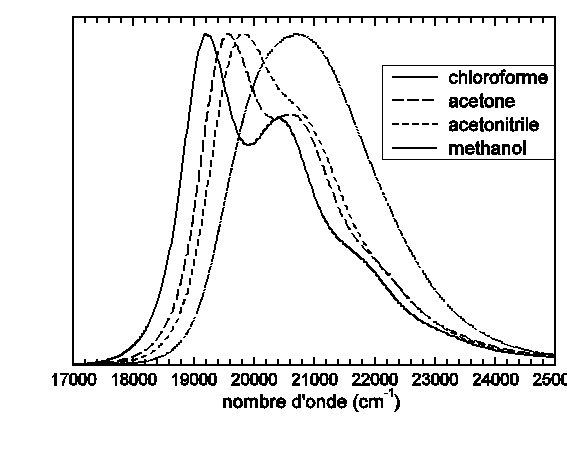
Spectre d’absorption UV-visible de la molécule de para-Méthyle-Pyridinium-Phenoxyde substituée de deux groupes tert-Butyle en ortho de l’oxygène dans différents solvants
Electric Field Induced Second Harmonic (EFISH) generation and Hyper-Rayleigh Scattering (HRS) experiments have so far been performed mostly in acetonitrile. Remarkably, these NLO results can be interpreted on the basis of semi-empirical AM1 calculations provided the COSMO solvation model is used while the solvatochromic shifts are not satisfactorily described. Concerning infrared (IR) and Raman spectroscopies, DFT based methods provide helpful results concerning the normal mode analysis but show limitations in describing geometrical changes with solvent polarities in continuum salvation models.
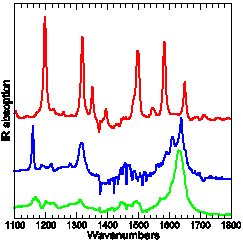
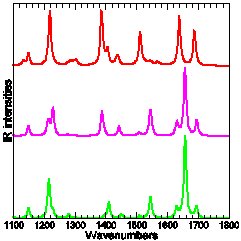
Experimental and calculated (DFT-B3LYP) IR absorption of three para-Methyl-Pyridinium-Phenoxide compounds: only the top-most carries tert-Butyl protection groups.
Key problem areas of organic NLO compounds such as solubility, aggregate formation, hydrogen bonding, or even protonation can also be studied with these molecules. In this respect, the tert-butyl protection groups have also demonstrated their role in reducing aggregation and in enhancing solubility. We hope that some of the results obtained in solutions will also prove useful in identifying problematic situations of chromophores in real application matrices such as polymeric media, for instance. Recently, in a large collaborative effort between Italy and the United States (JACS 2007, 129, 3267), tictoid (twisted intramolecular charge transfer) structures achieved through inter-aryl sterical crowding leading to very large twist angles have been claimed to exhibit hyperpolarizabilities that are between two and four hundred times larger than those obtained with our less twisted biaryls. Although a stronger electron acceptor group carrying two nitrile functions has been used in this instance in conjunction with the pyridinium donor group, analogous predictions have been made for the oxygen capped variety in semi-empirical, density functional theory (DFT), and standard ab initio calculations performed mostly in the gas phase. Calculations also pointed towards very low lying electronic excited states, but these have not yet been confirmed experimentally. Also, the geometries of these tictoid structures appear to remain mainly unchanged between the crystalline state and in solutions, while their hyperpolarizabilities have been found to strongly depend on the solvent in more recent electronic structure calculations that included solvation effects. Interestingly, these observations stand in contrast to those we have made about our less twisted biaryls. Obviously, there remain many issues to be settled, both experimentally and theoretically, before we will be able to claim to fully understand these molecular systems.